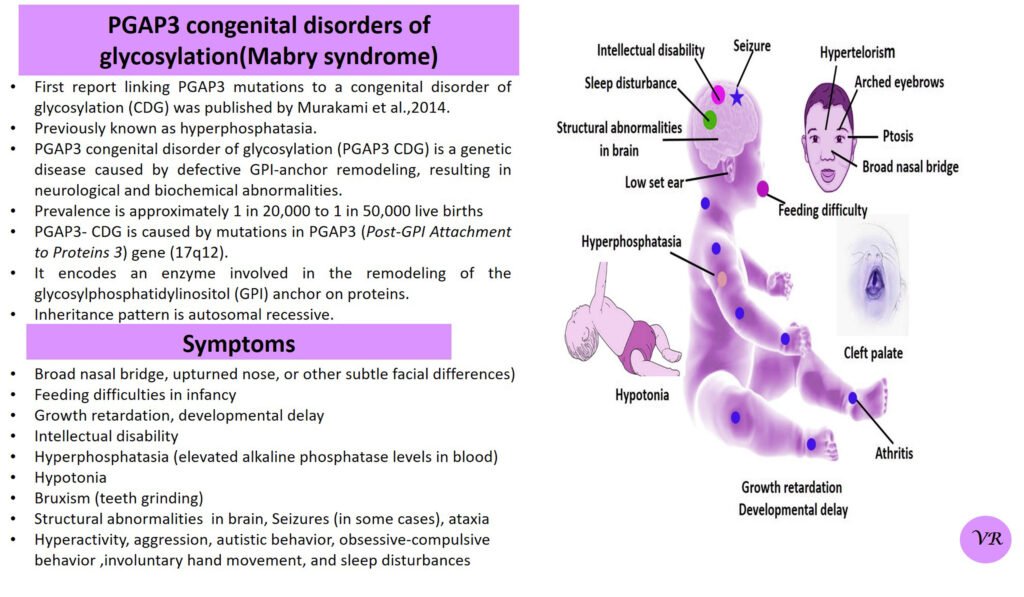
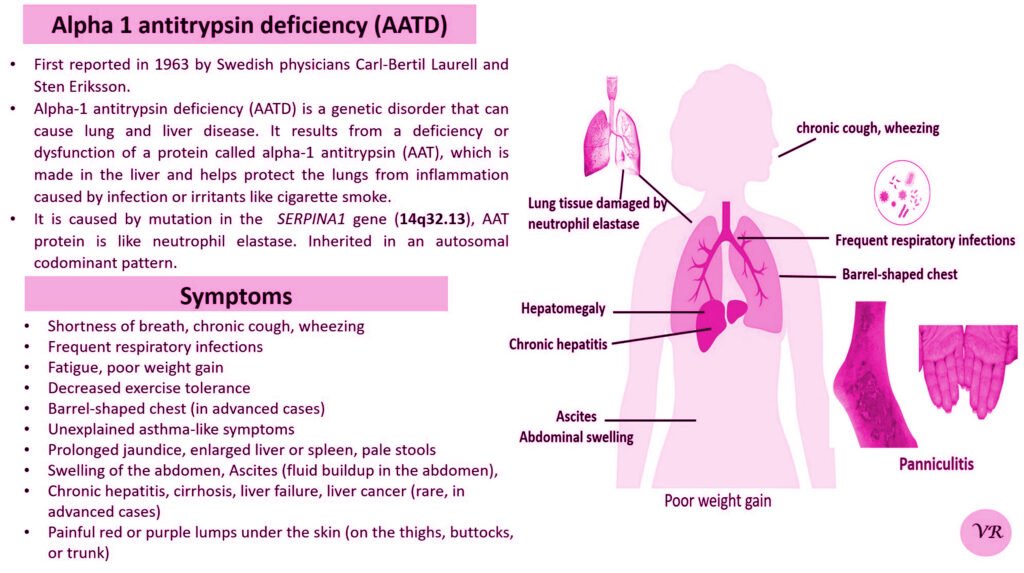
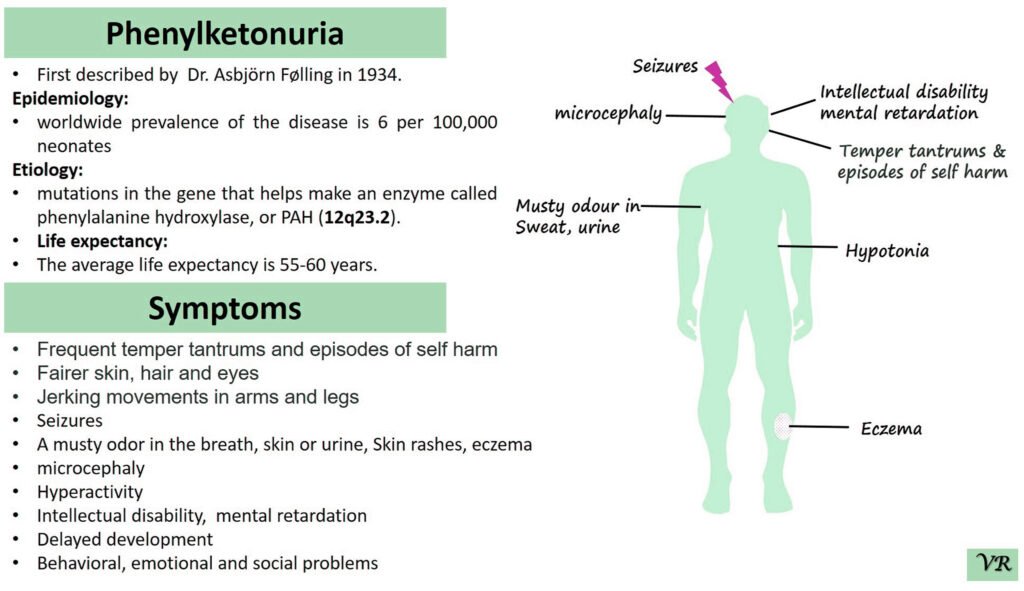
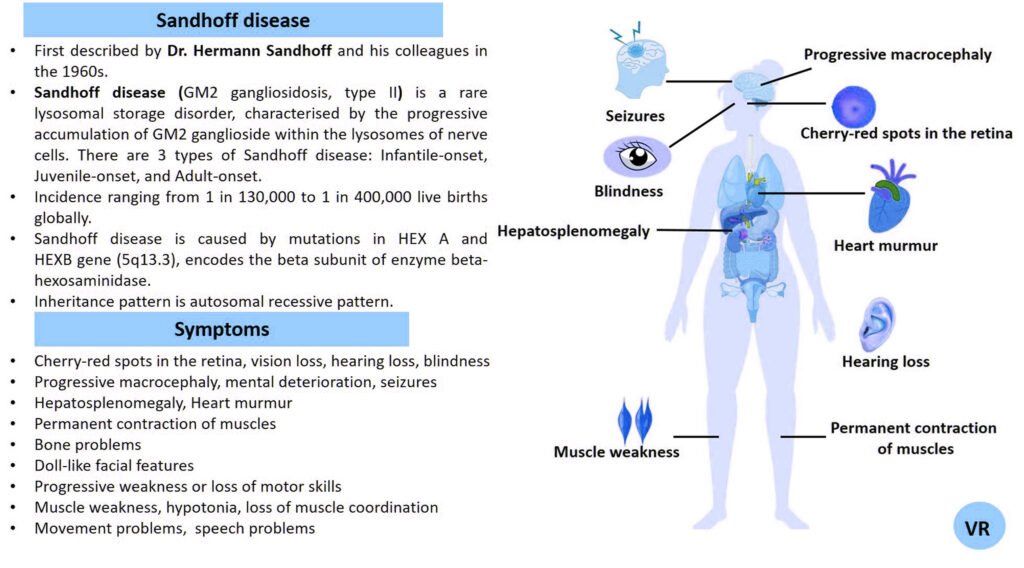
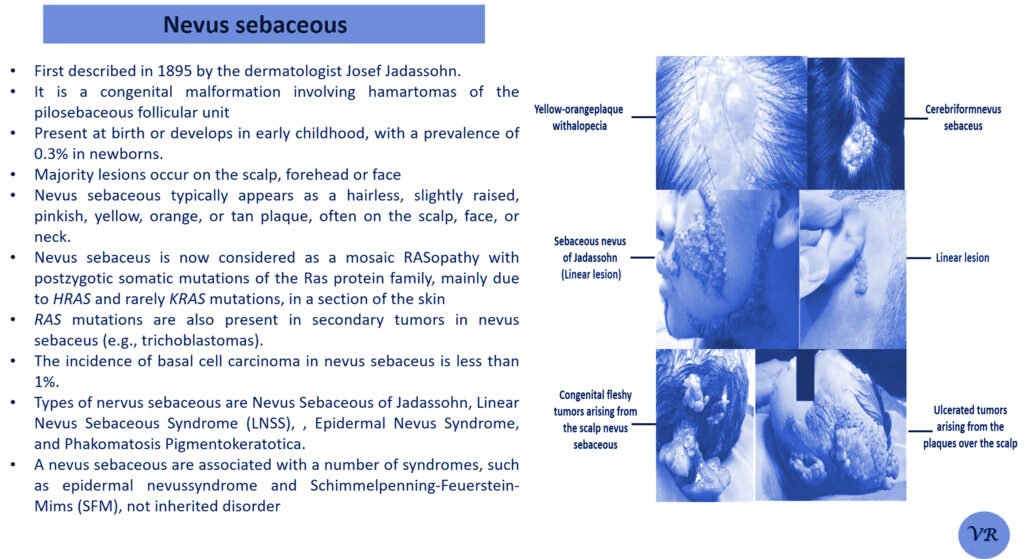
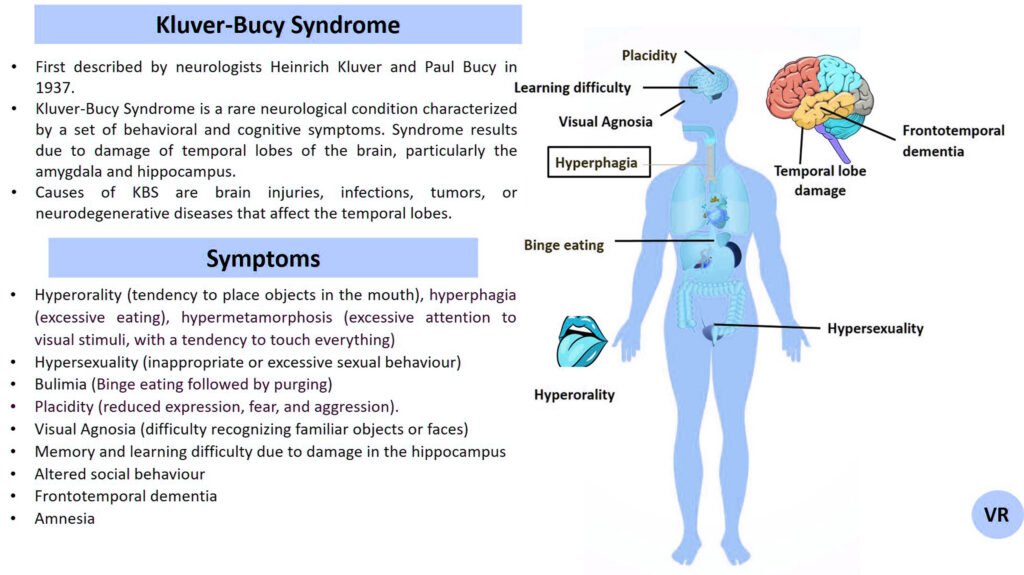
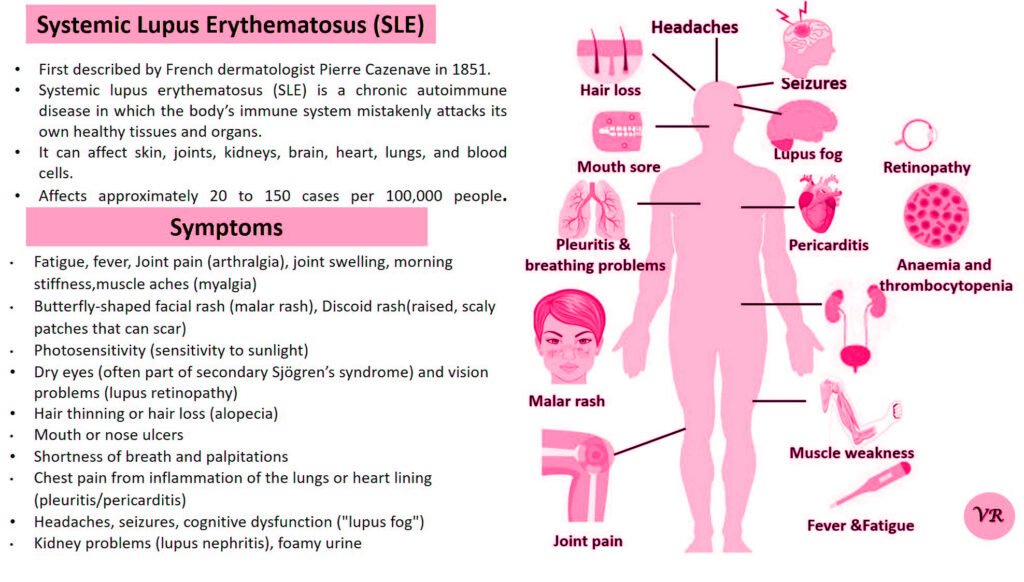
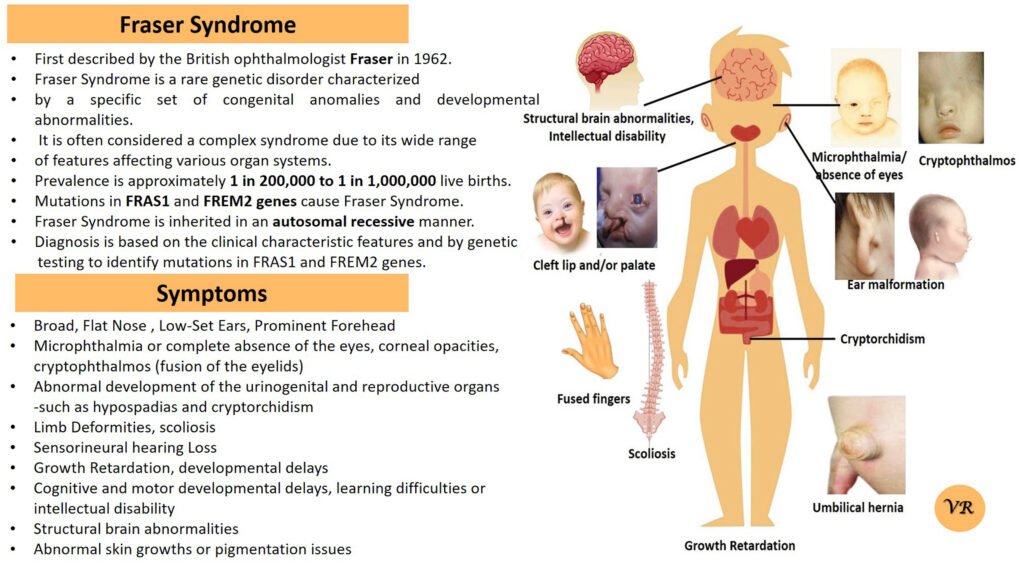
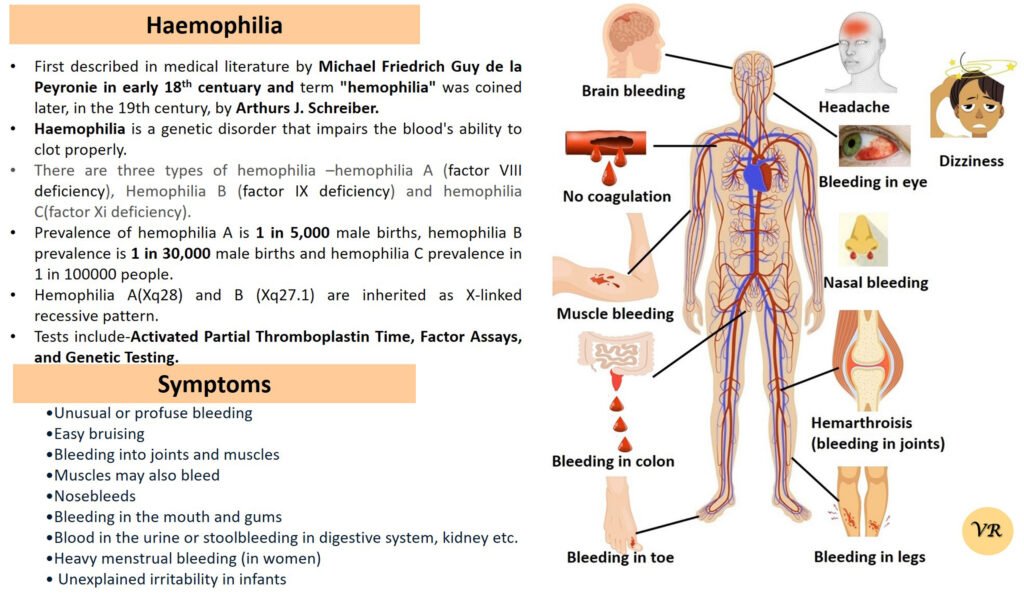
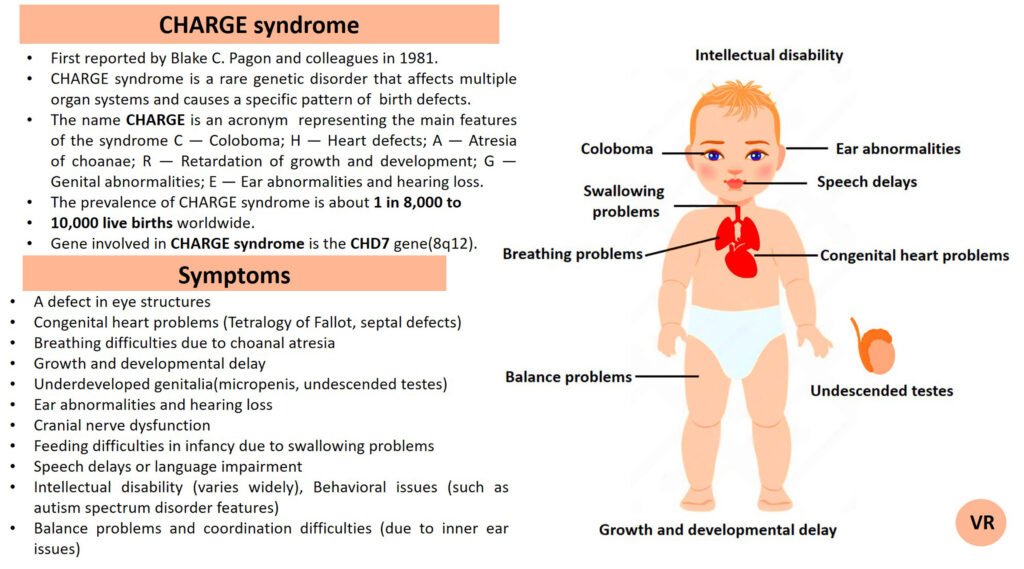
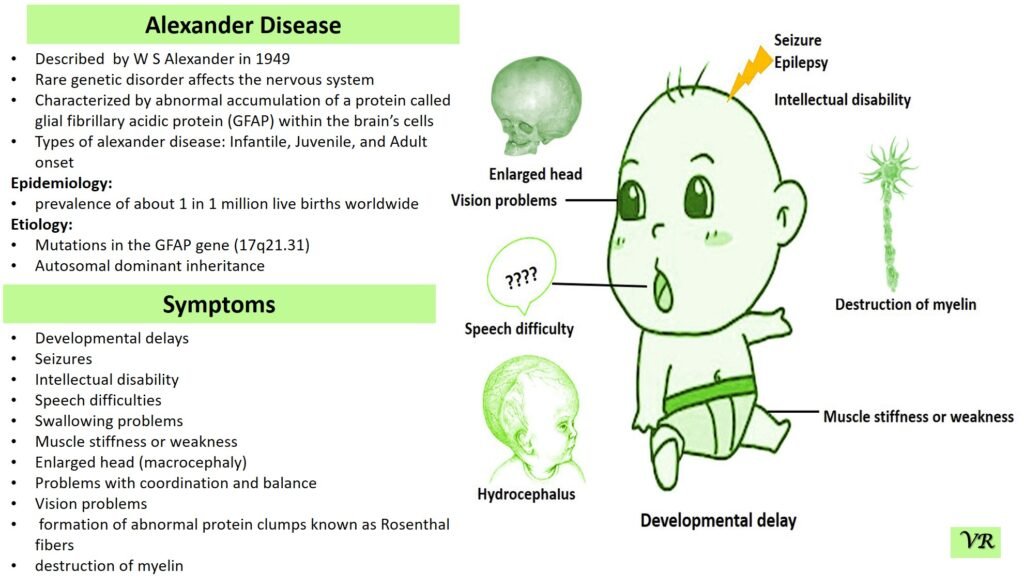
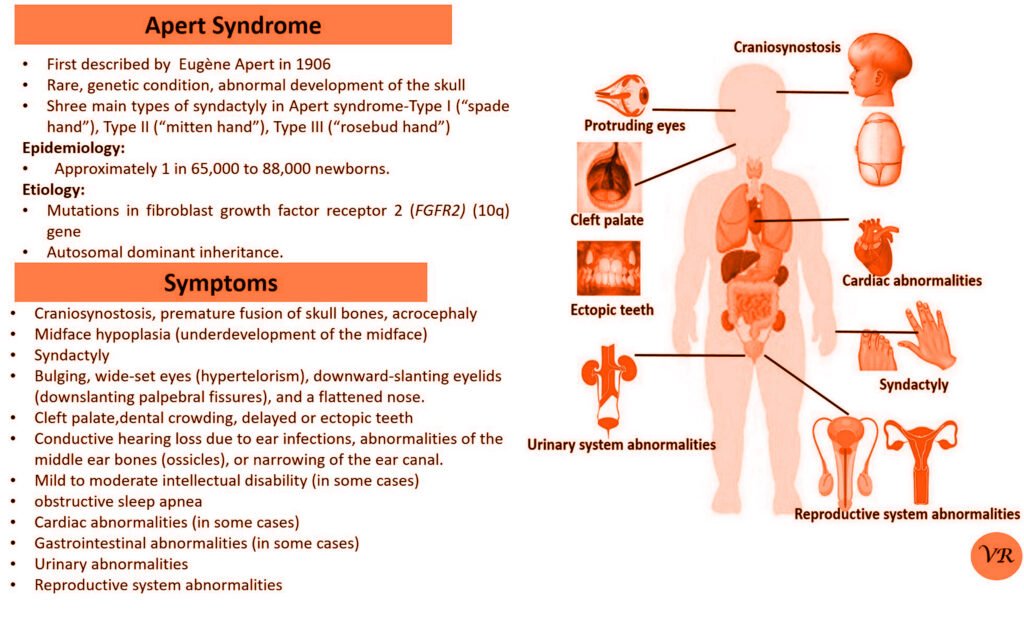
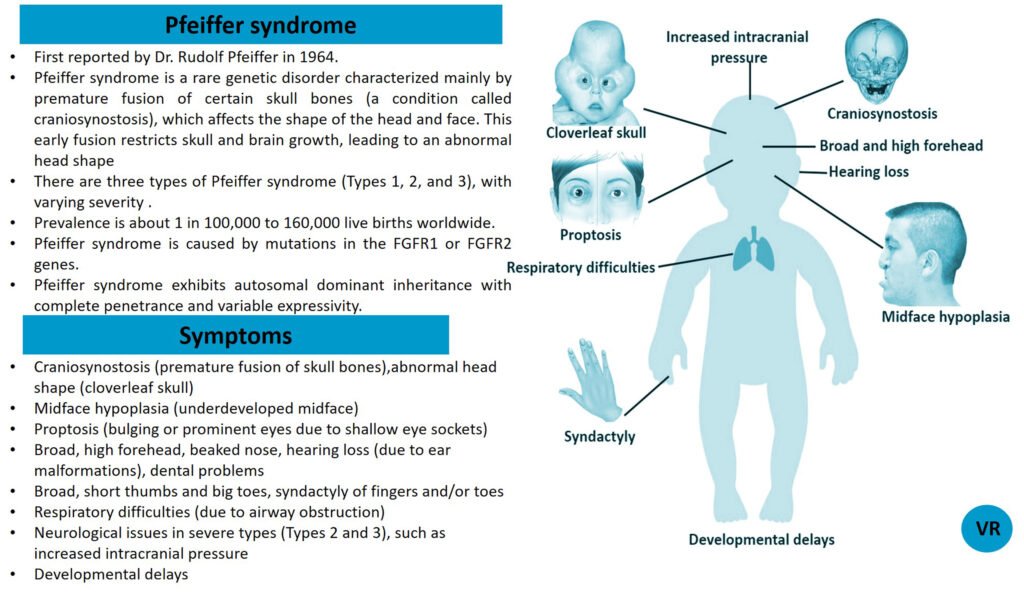
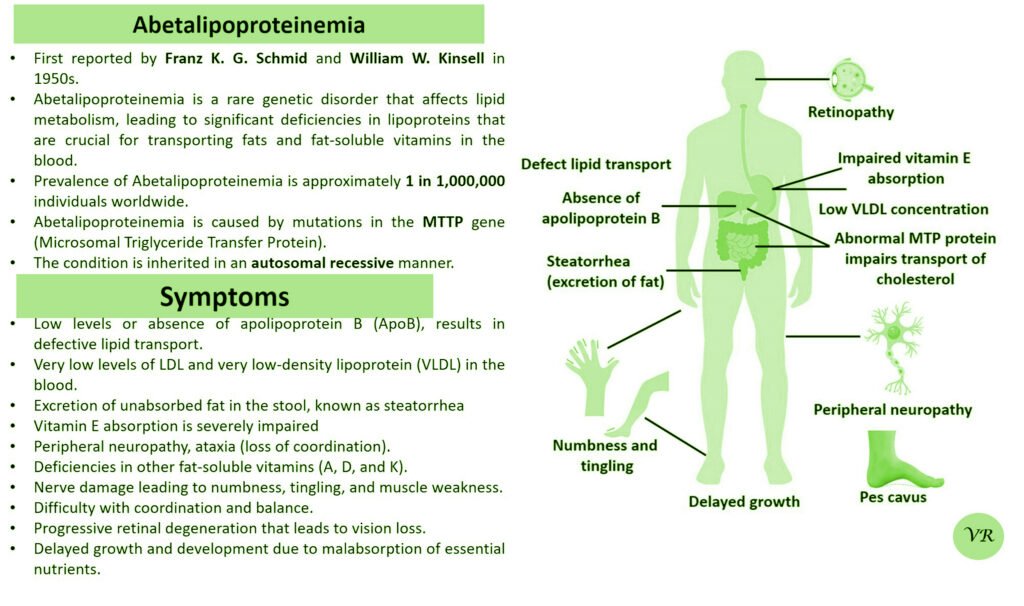
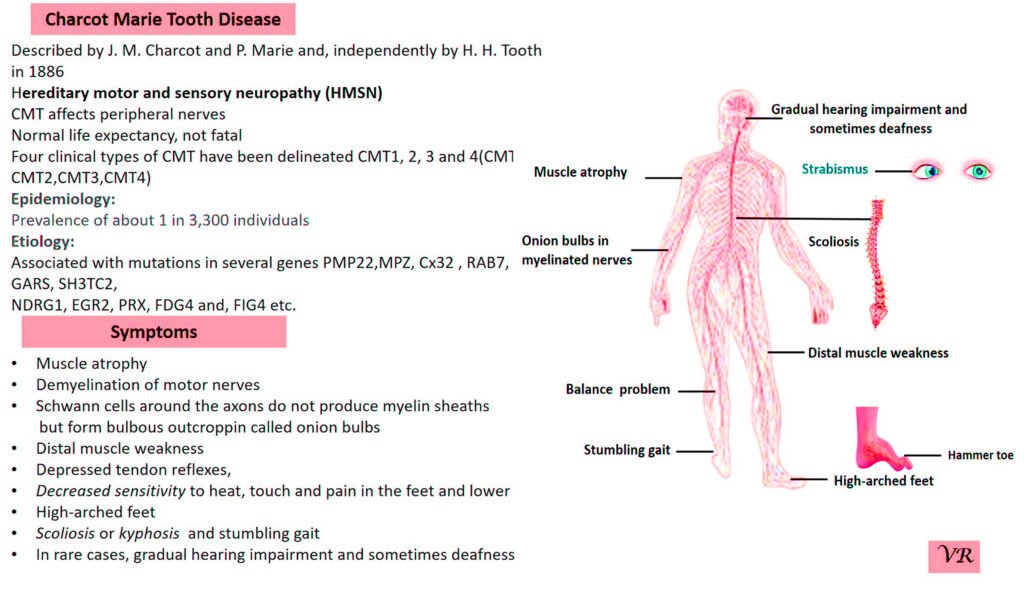
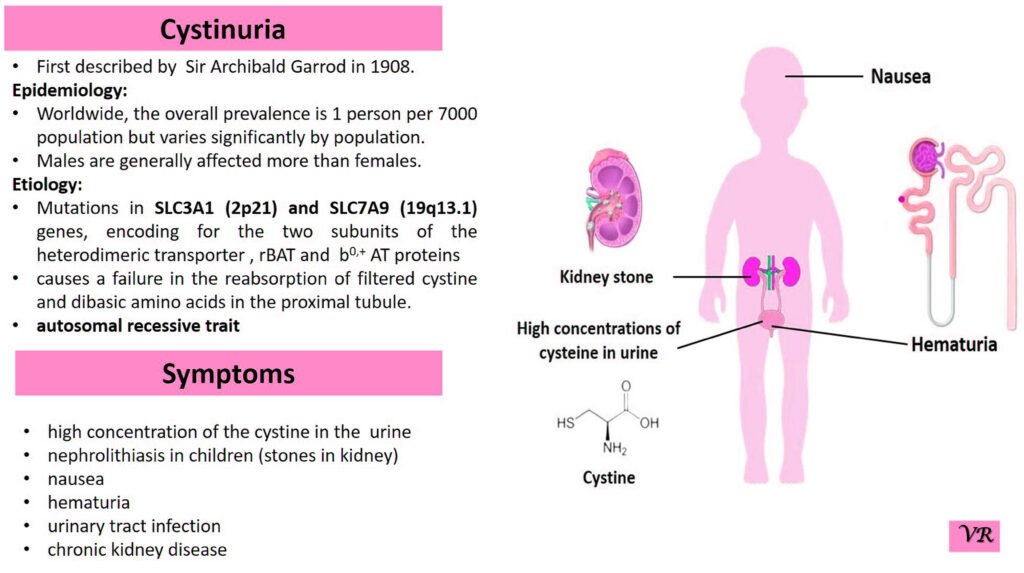
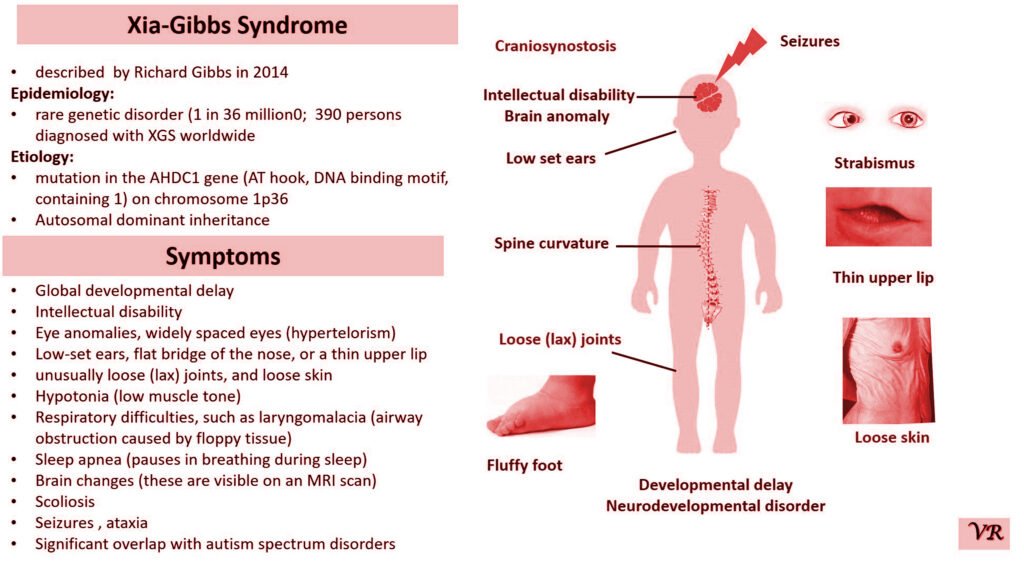
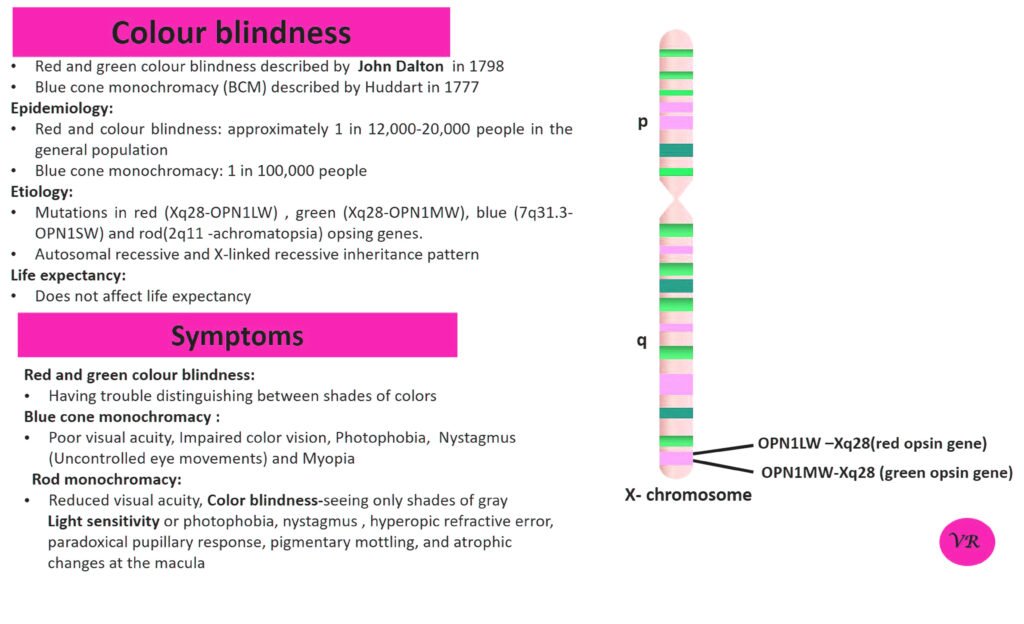
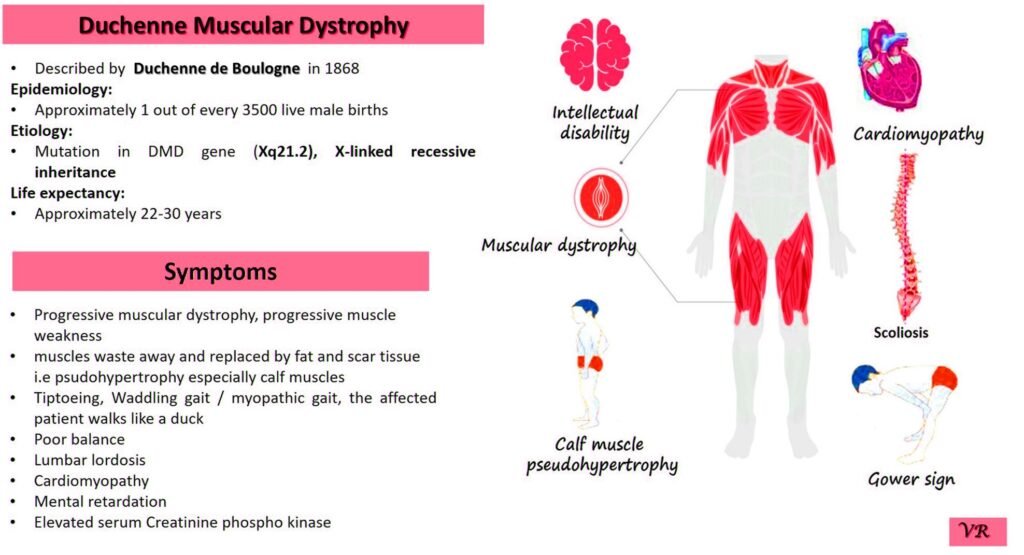
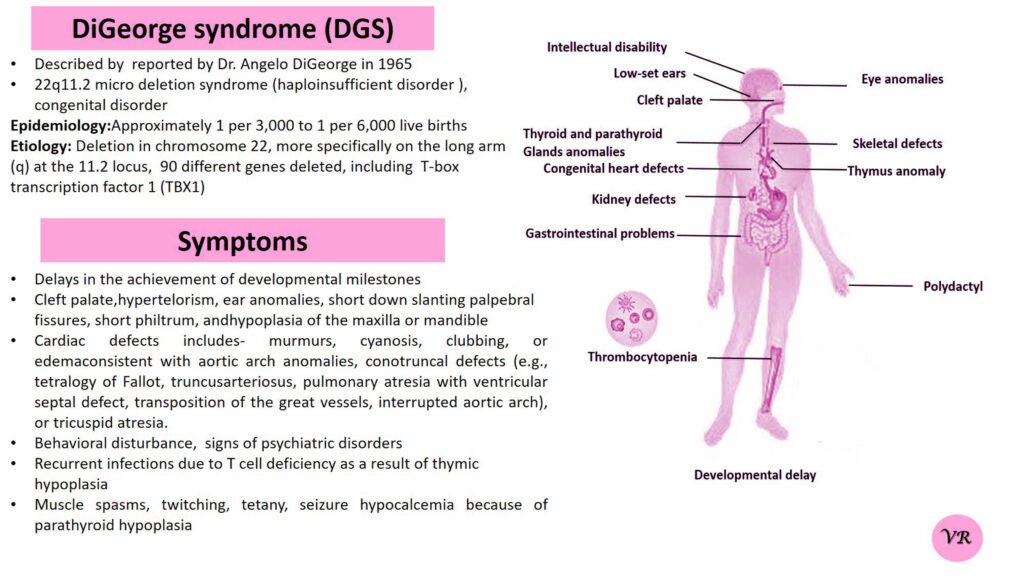
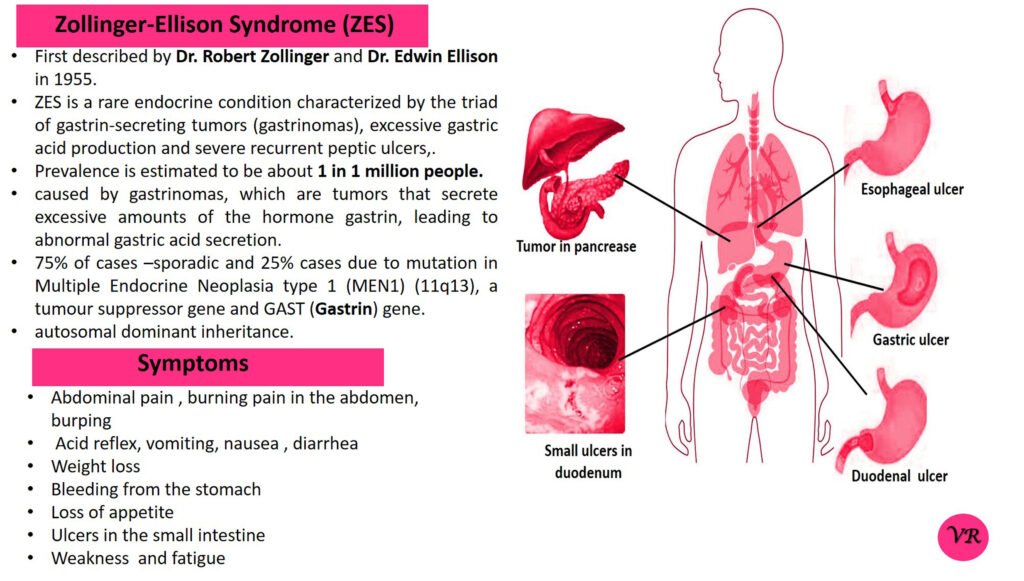
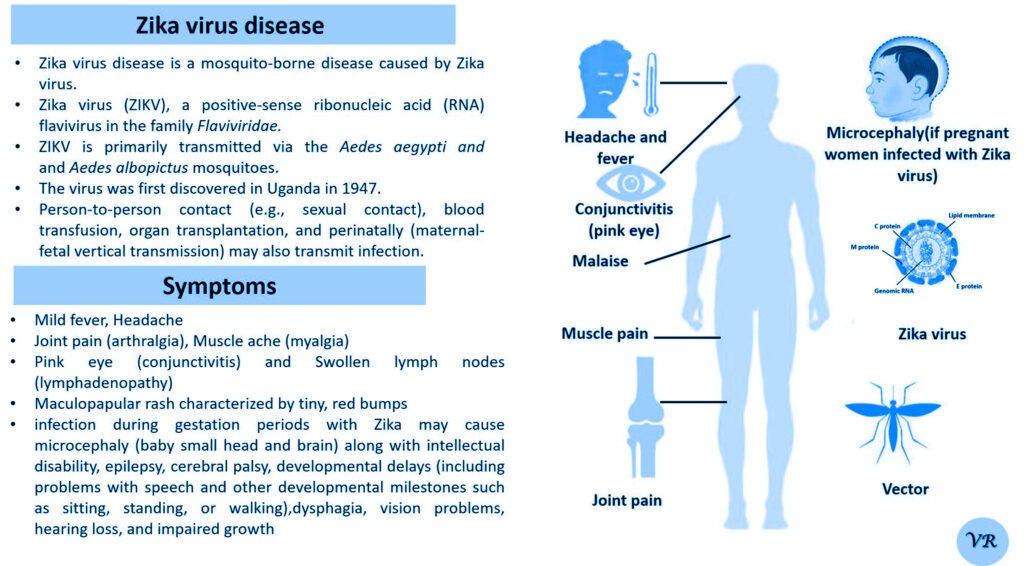
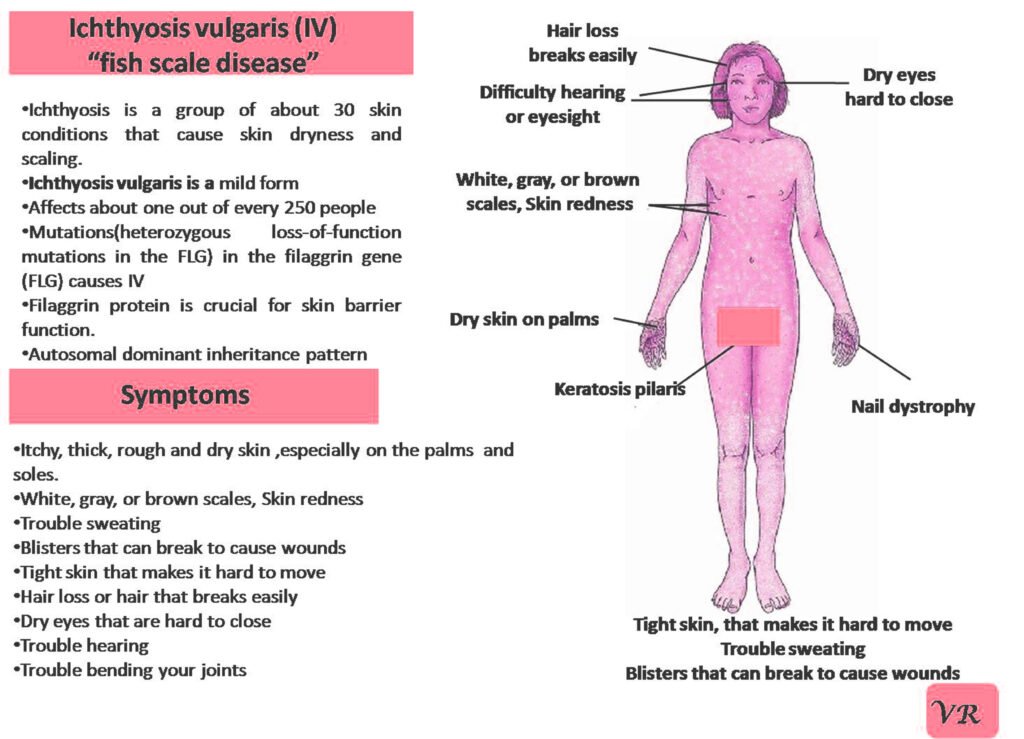
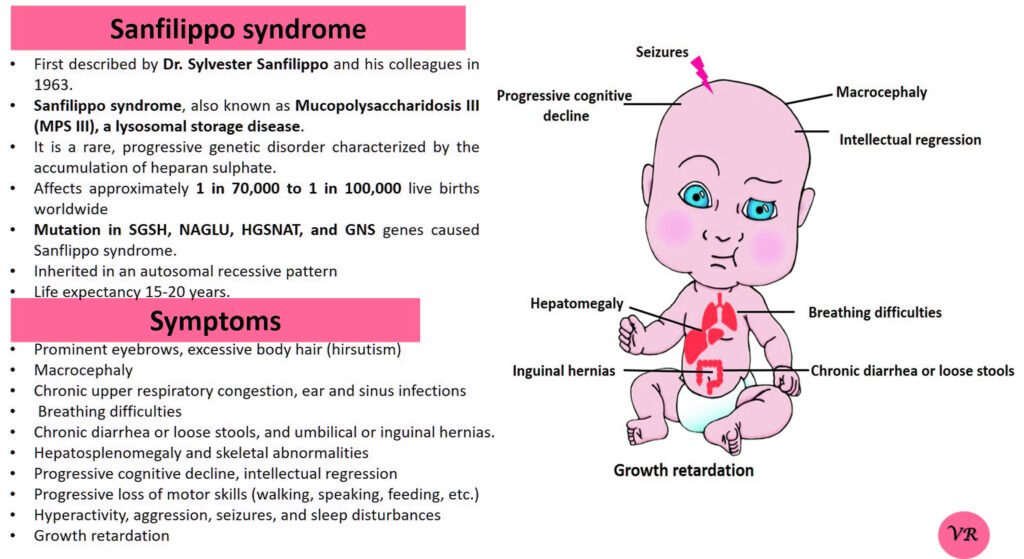
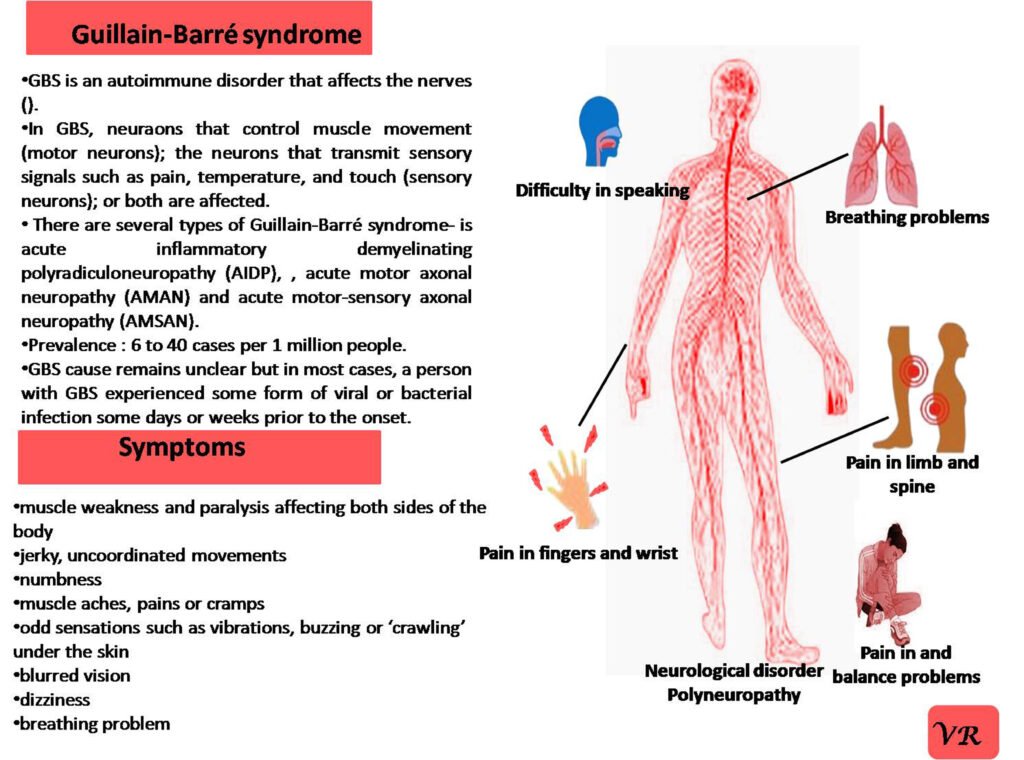
Phenylketonuria (PKU) is a genetic metabolic disorder characterized by the inability to metabolize the amino acid phenylalanine due to a deficiency in the enzyme phenylalanine hydroxylase and phenylalanine cannot be effectively converted to tyrosine. This results in the accumulation of phenylalanine in the blood and is converted into phenylpyruvate and other metabolites which can be harmful, especially to the brain cause intellectual disability and other neurological problems. Prevalence rate is 1 in 10,000 to 15,000 live births worldwide. Symptoms includes intellectual disability, behavioral problems and psychiatric symptoms, eczema, seizures, and musty odour in the breath, skin, and urine due to the accumulation of phenylketones.
PKU is caused by mutations in the phenylalanine hydroxylase (PAH) gene, which encodes the enzyme phenylalanine hydroxylase. This enzyme is responsible for converting phenylalanine into tyrosine. Without this enzyme, phenylalanine accumulates and leads to toxic effects.
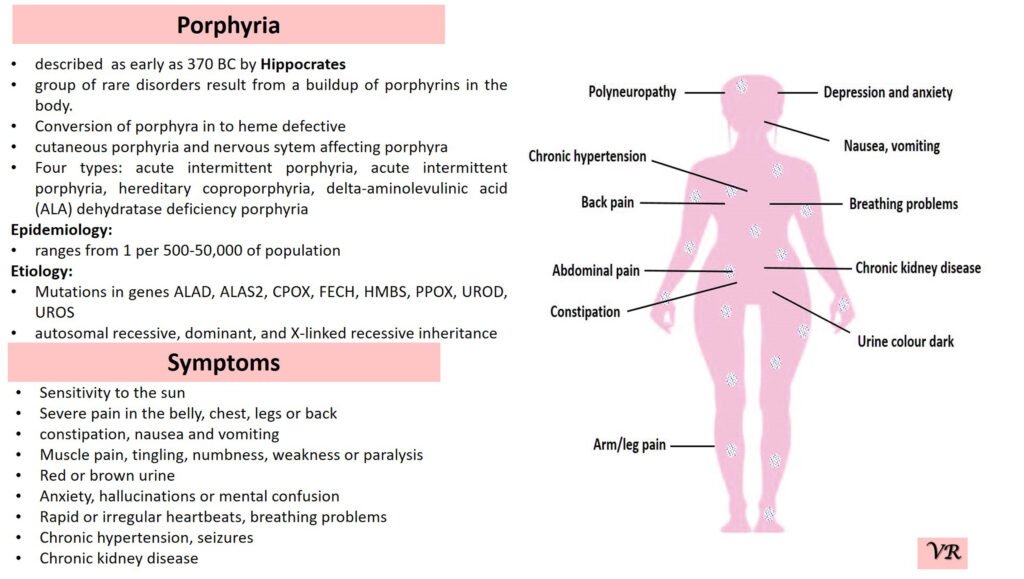
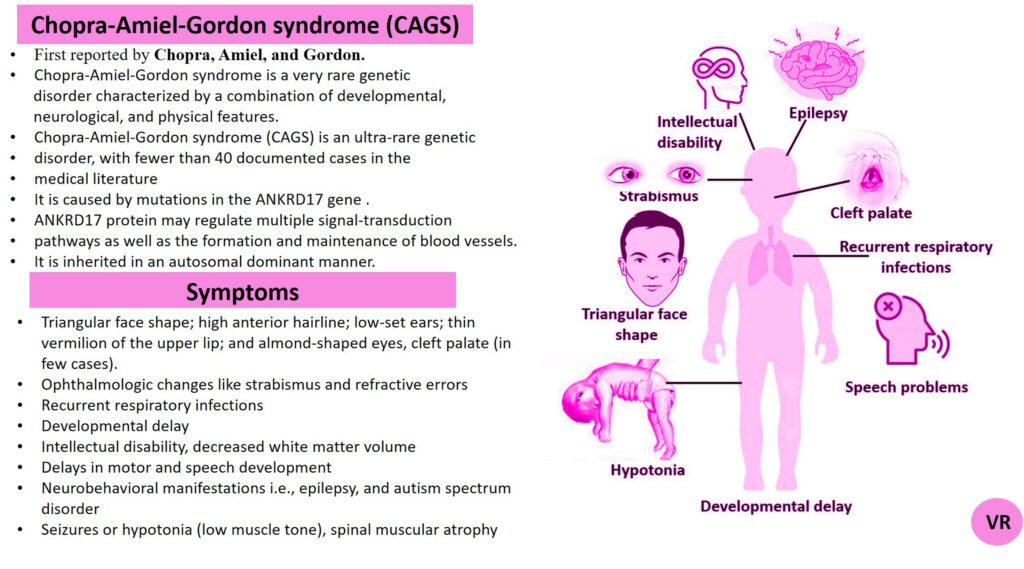
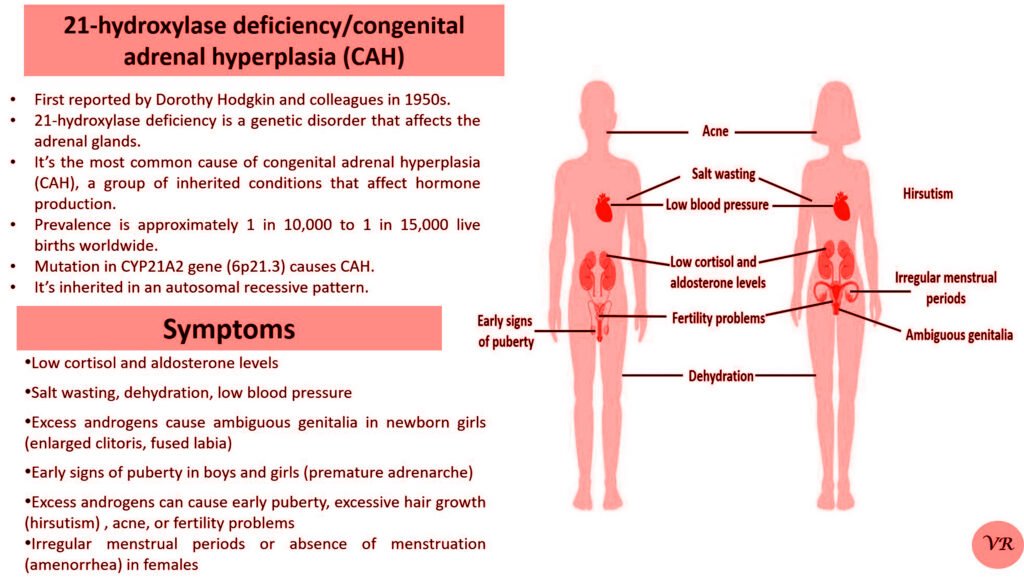
Porphyria
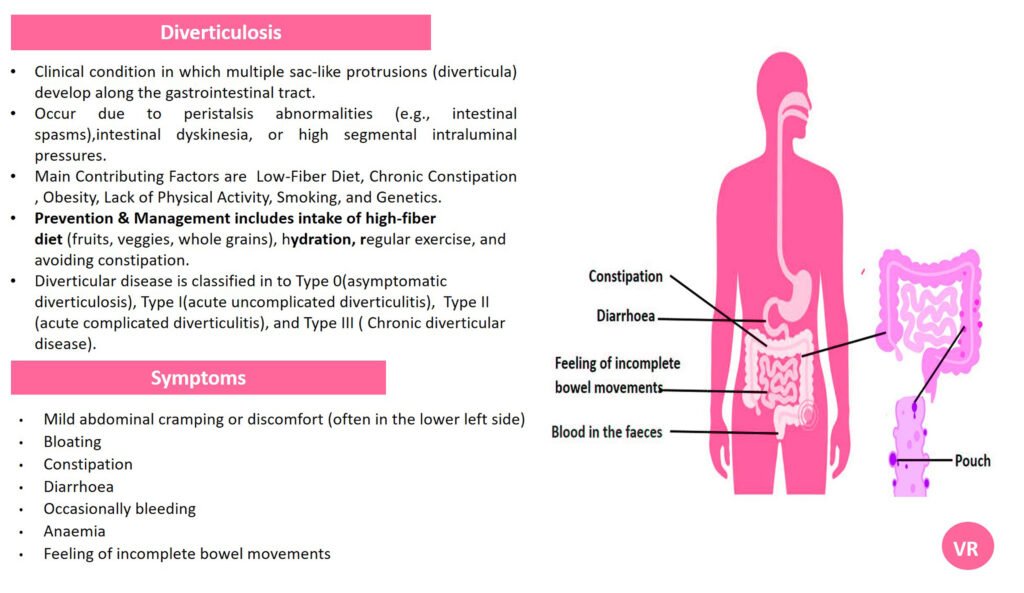
Porphyria is a group of rare metabolic disorders caused by defects in the heme biosynthesis pathway, leading to the accumulation of porphyrins or their precursors. It can present with a range of symptoms, including abdominal pain, neurological disturbances, and skin sensitivity to sunlight. Prevalence of porphyria is 1 in 25,000 to 50,000 individuals worldwide. Several specific types of porphyria are reported such as (i)Acute Intermittent Porphyria (AIP), Porphyria Cutanea Tarda (PCT), Hereditary Coproporphyria (HCP), Variegate Porphyria (VP), and Erythropoietic Protoporphyria (EPP). Each type of porphyria is associated with mutations in specific genes that encode enzymes involved in this pathway. Here are the key genes involved in different types of porphyria- Hydroxymethylbilane Synthase gene (Acute Intermittent Porphyria), Uroporphyrinogen Decarboxylase gene (Porphyria Cutanea Tarda), Coproporphyrinogen Oxidase gene (Hereditary Coproporphyria), Protoporphyrinogen Oxidase gene (Variegate Porphyria), and Ferrochelatas gene (Erythropoietic Protoporphyria).
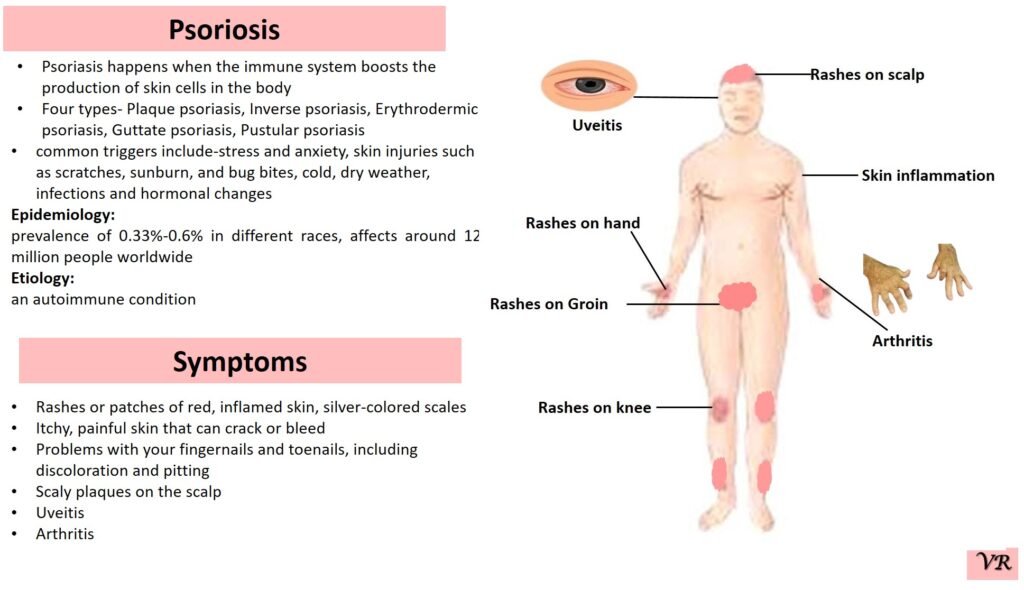
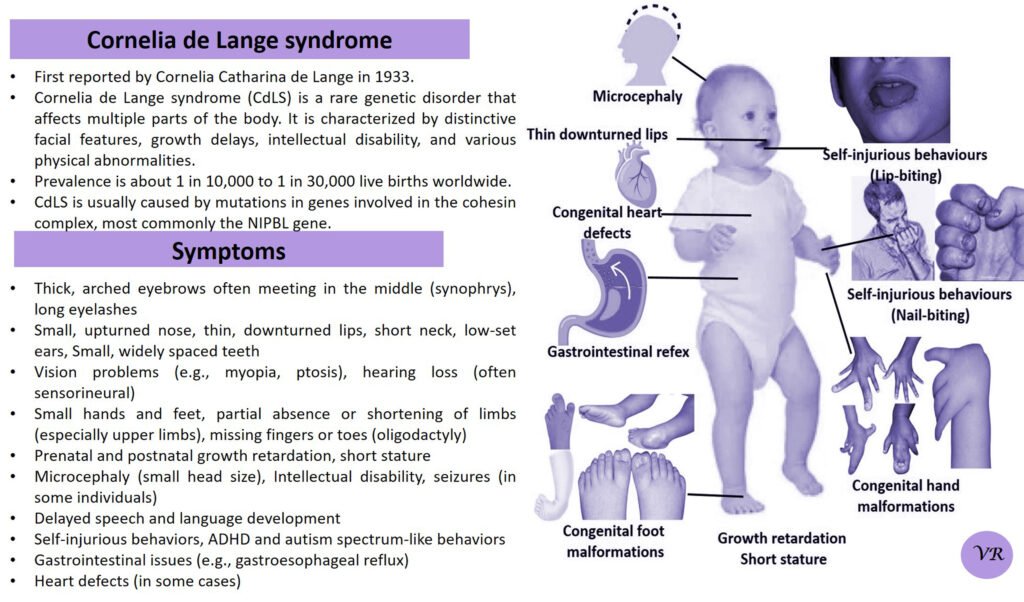
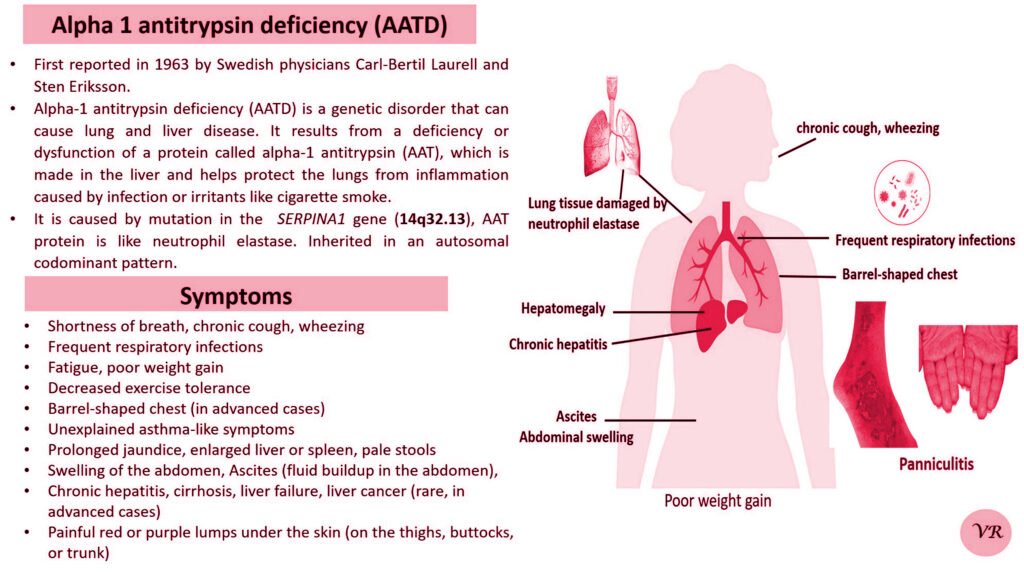
Psoriasis
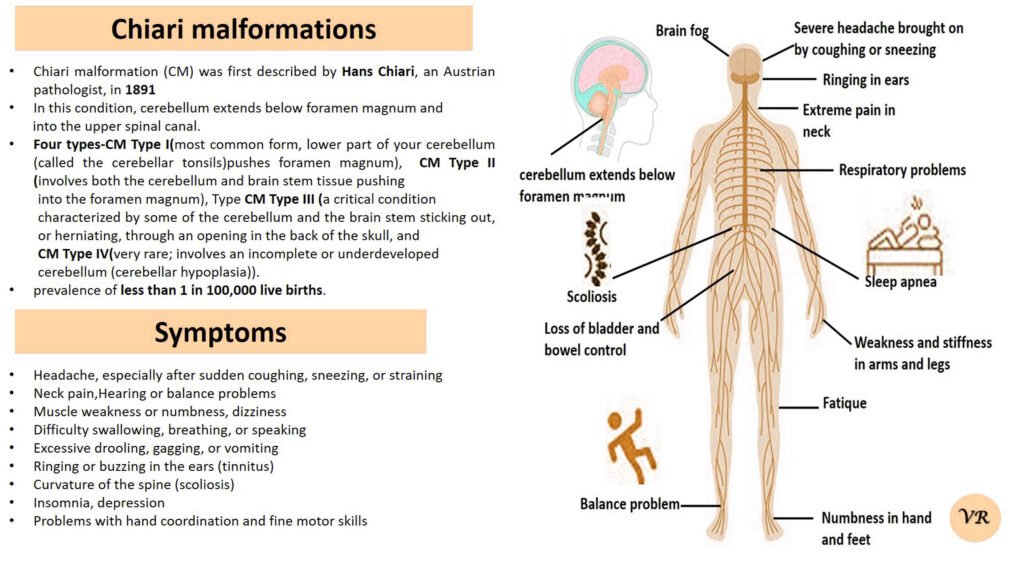
Psoriasis is a chronic autoimmune skin condition that causes rapid skin cell growth, leading to thick, red, scaly patches on the skin. It is a common condition that can vary in severity and impact different areas of the body. The earliest descriptions of psoriasis date back to ancient times, but the condition was formally recognized and described in more detail in the 19th century. Prevalence is 1% to 3% of the global population. The symptoms can vary depending on the type of psoriasis, but common signs include patches of red skin covered with thick, silvery-white scales, typically found on elbows, knees, scalp, and lower back, dry cracked skin, itching or burning sensation, thickened or ridged nails, swollen or stiff joints and shedding of the skin that and can lead to serious health complications if not treated.
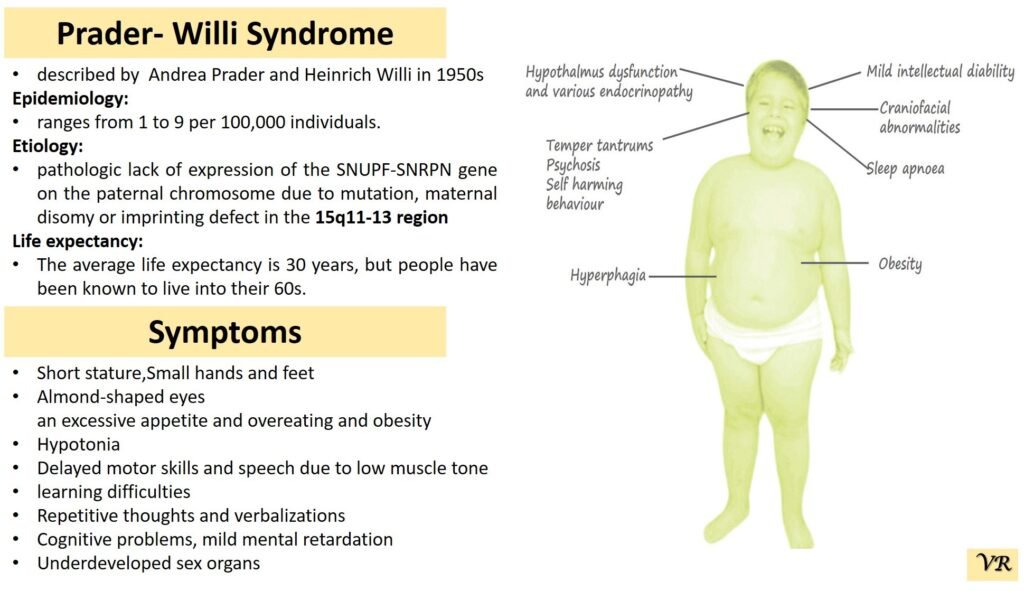
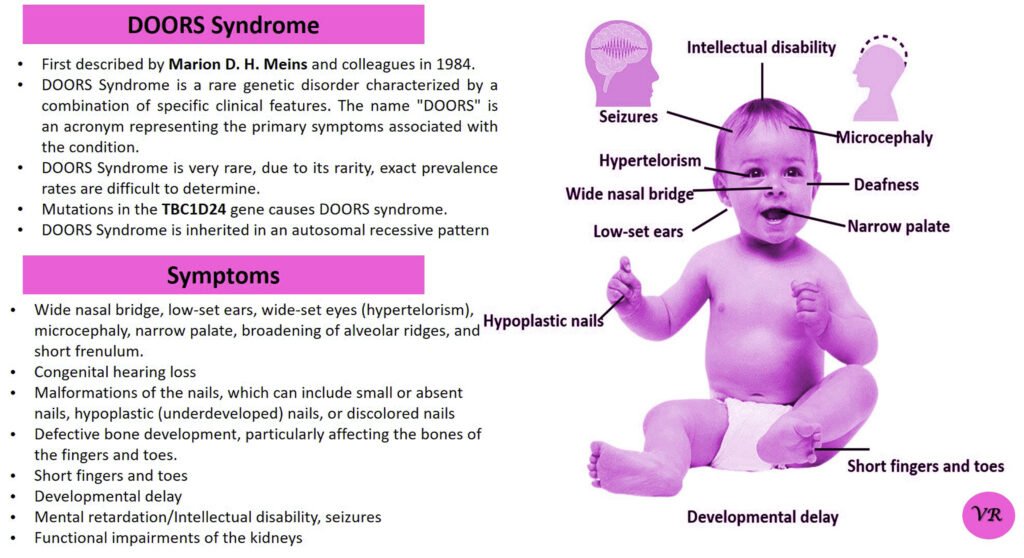
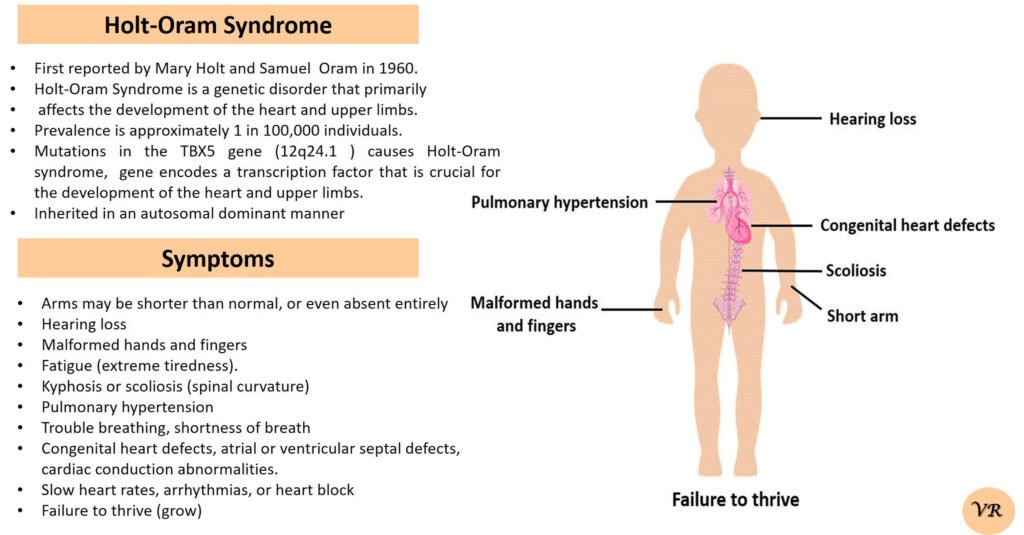
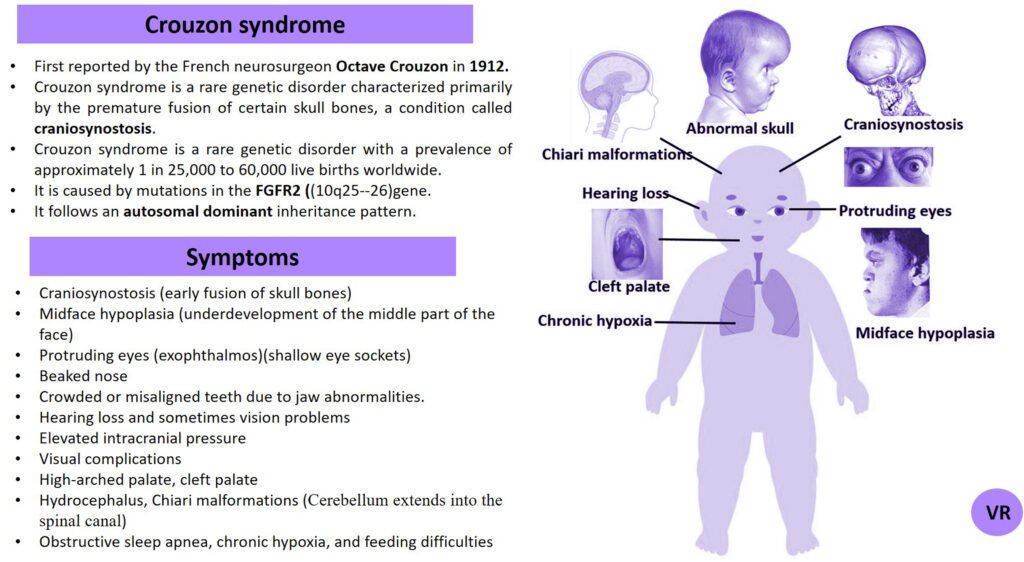
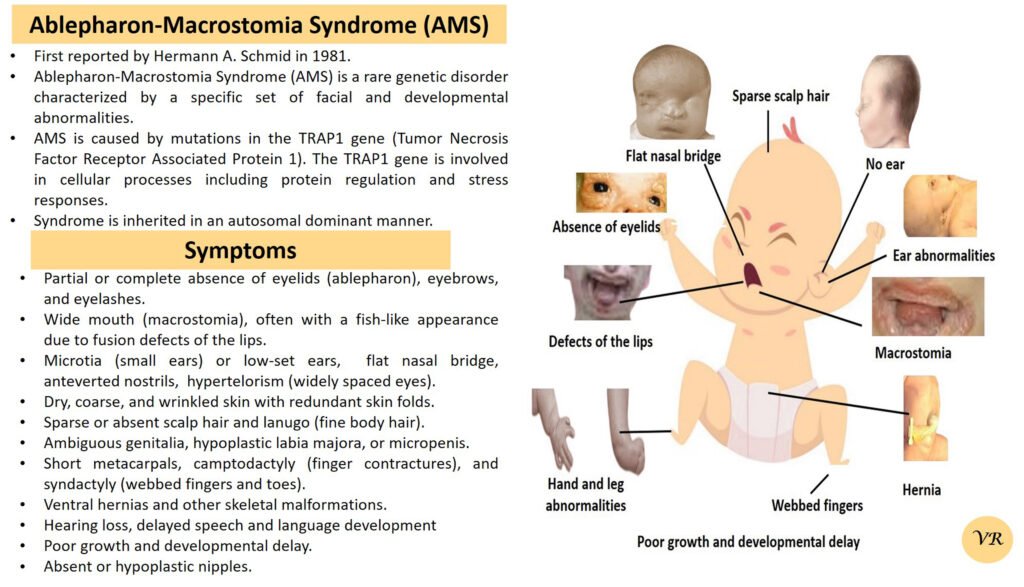
Prader-Willi syndrome
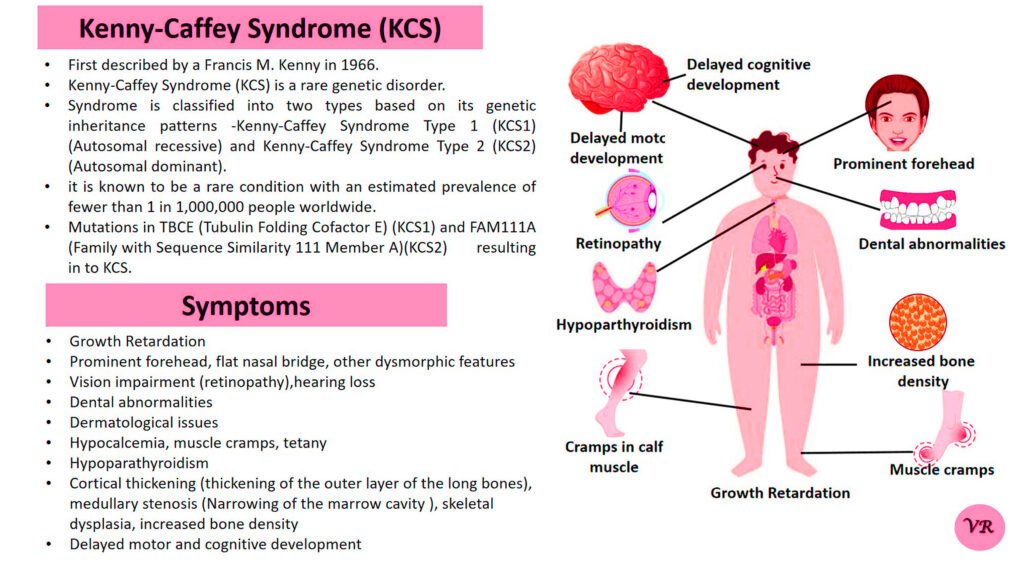
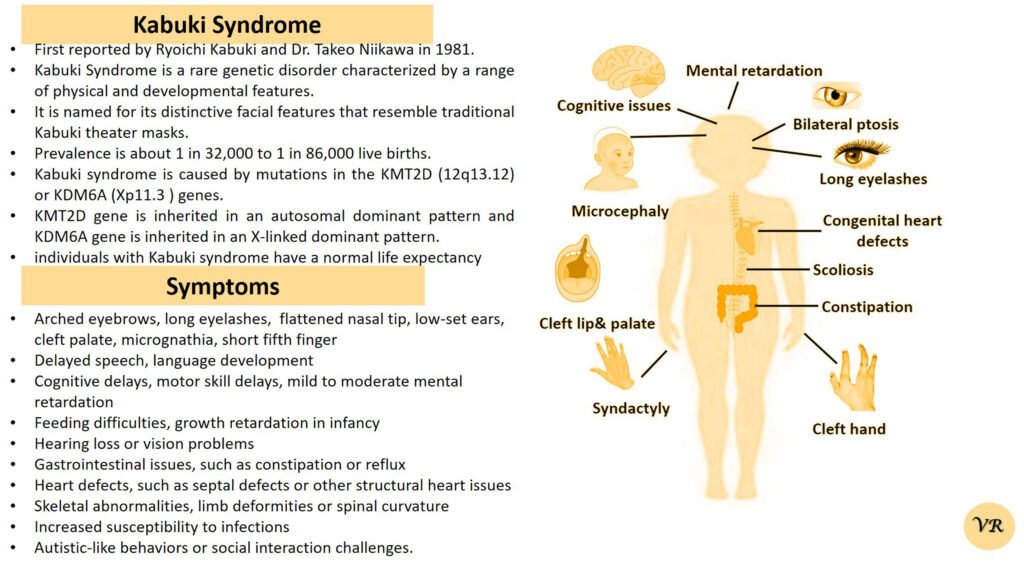
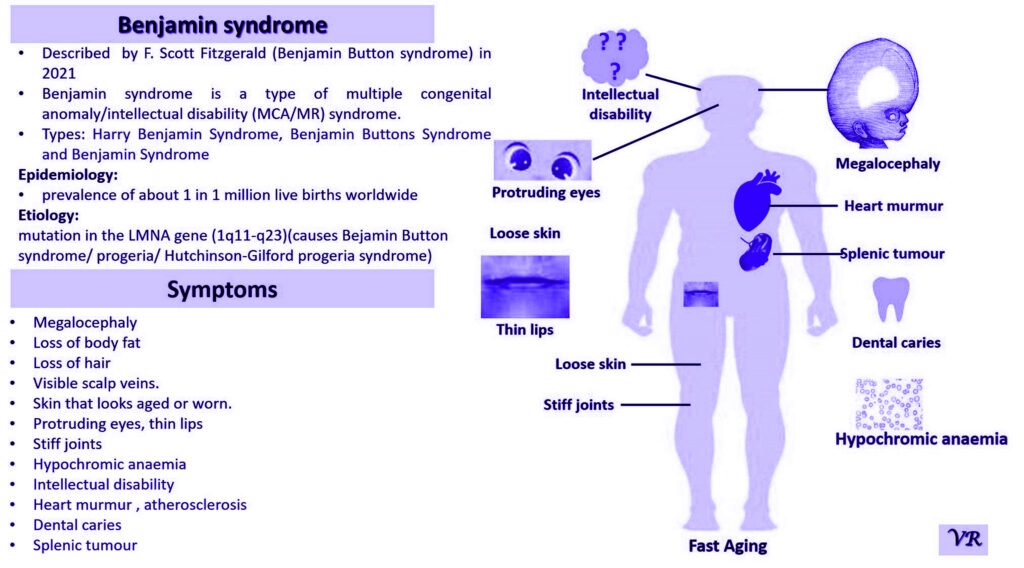
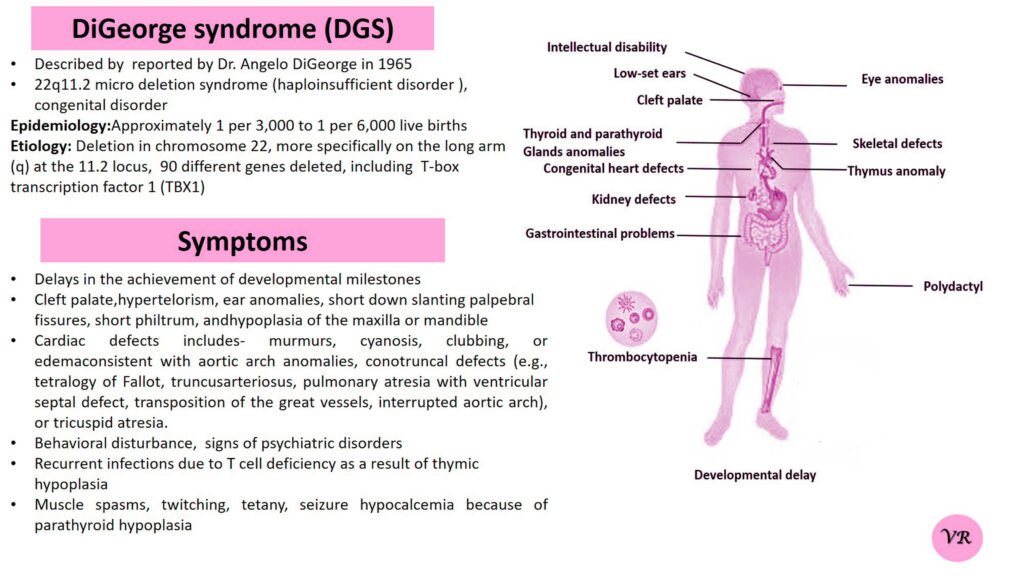
Prader-Willi Syndrome (PWS) is a complex genetic disorder that affects multiple aspects of physical and cognitive development. It is characterized by a combination of symptoms that include intellectual and developmental disabilities, obesity, and various hormonal and behavioral issues. 1 in 10,000 to 15,000 live births worldwide. Symptoms include short stature, narrow forehead, prominent chin, hypotonia, feeding problems, hyperphagia, obesity, type 2 diabetes, growth hormone deficiency, intellectual disability, behavioral issues (temper tantrums, stubbornness, and obsessive-compulsive behaviors), sleep apnea, and hypogonadism (reduced function of the sex glands leads to incomplete or absent puberty). Prader-Willi syndrome is caused by the loss of function of genes on the paternal copy of chromosome 15, specifically in the 15q11-q13 region.
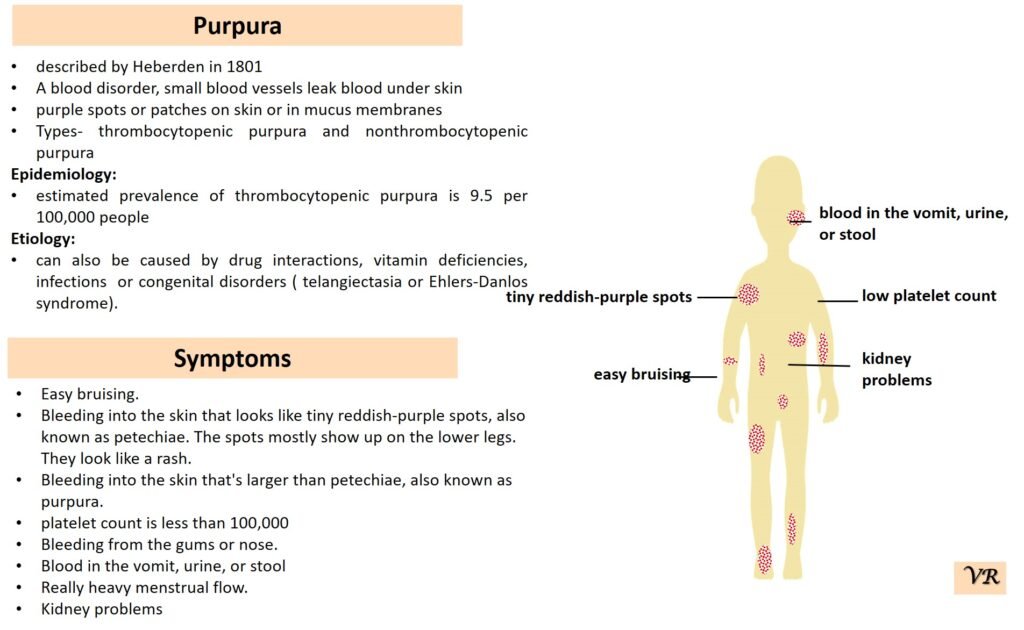
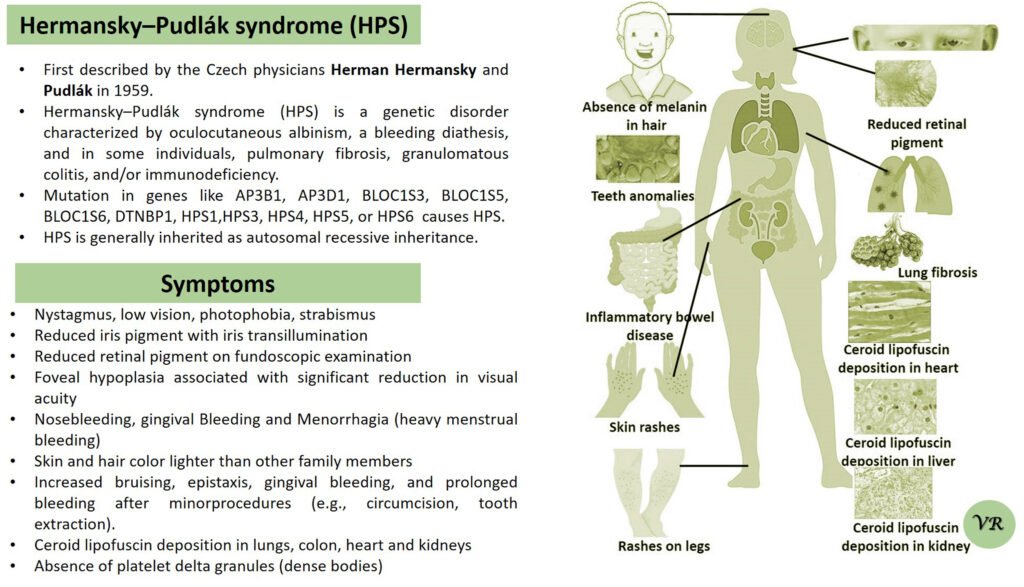
Purpura
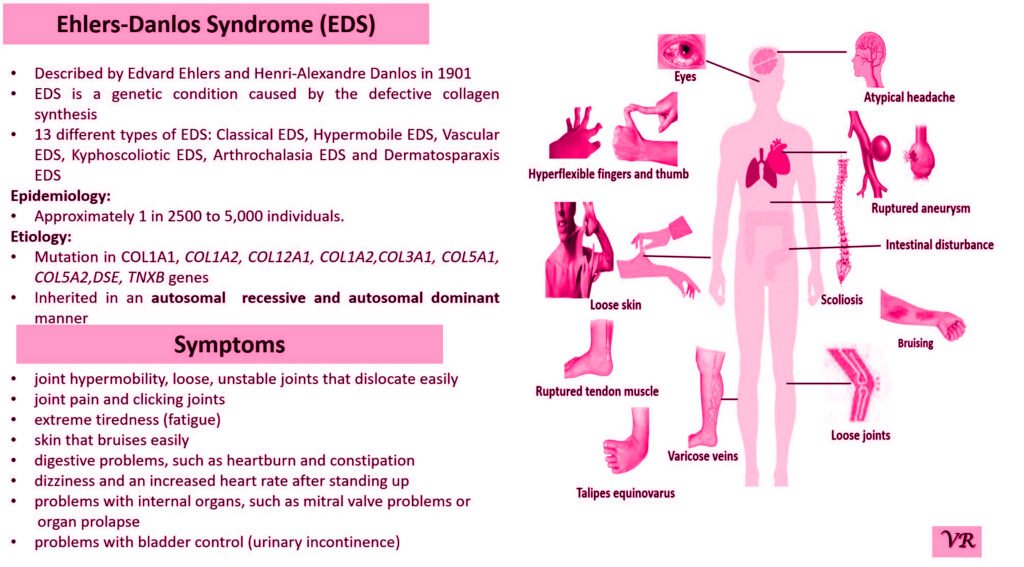
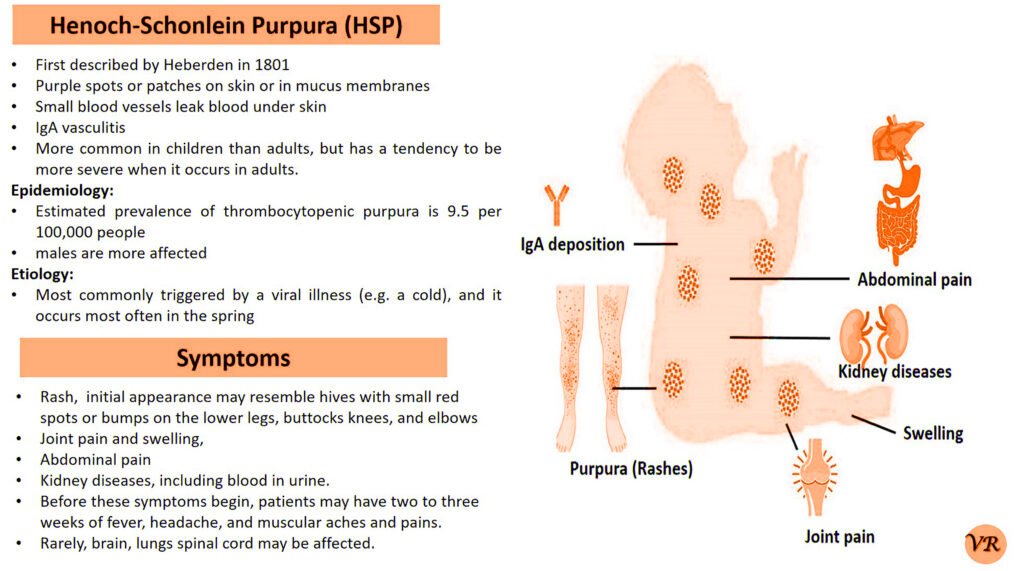
Purpura refers to a condition characterized by the appearance of purple or red spots and patches on the skin due to bleeding underneath the skin. It is a symptom rather than a specific disease and can result from various underlying conditions affecting blood vessels or blood clotting. The primary symptom is the appearance of purplish spots or patches on the skin. Depending on the underlying cause, purpura may be accompanied by other symptoms such as joint pain, bleeding gums, or easy bruising. Causes of purpura are vascular issues (vasculitis or scurvy), platelet disorders (such as thrombocytopenia or immune thrombocytopenic purpura), coagulation disorders (such as hemophilia or vitamin K deficiency), trauma or injury, and infections. Purpura itself is not directly caused by genetic factors, but it can result from underlying conditions that have genetic components. However, some key genes and genetic factors are involved in the conditions that commonly cause purpura: Factor VIII (F8) gene, Factor IX (F9) gene, Von Willebrand Factor gene, COL5A1 and COL5A2 genes, and Fibrillin-1 gene.
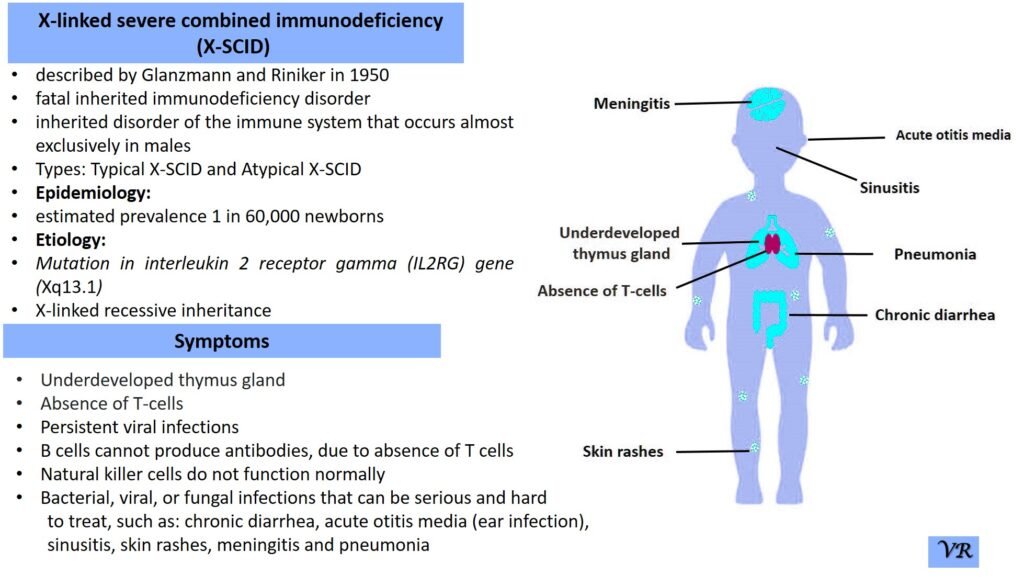
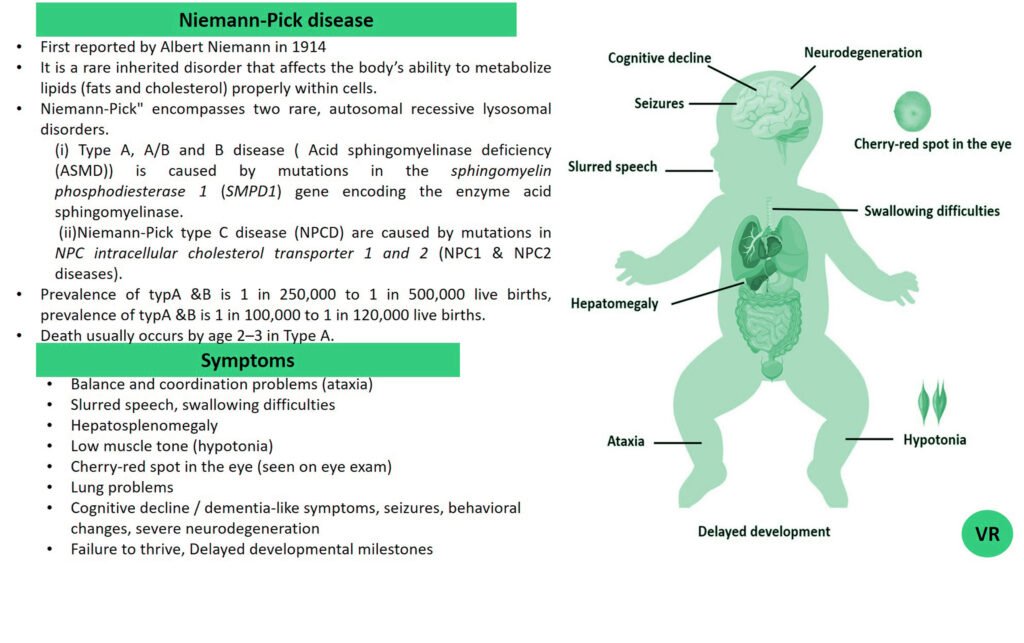
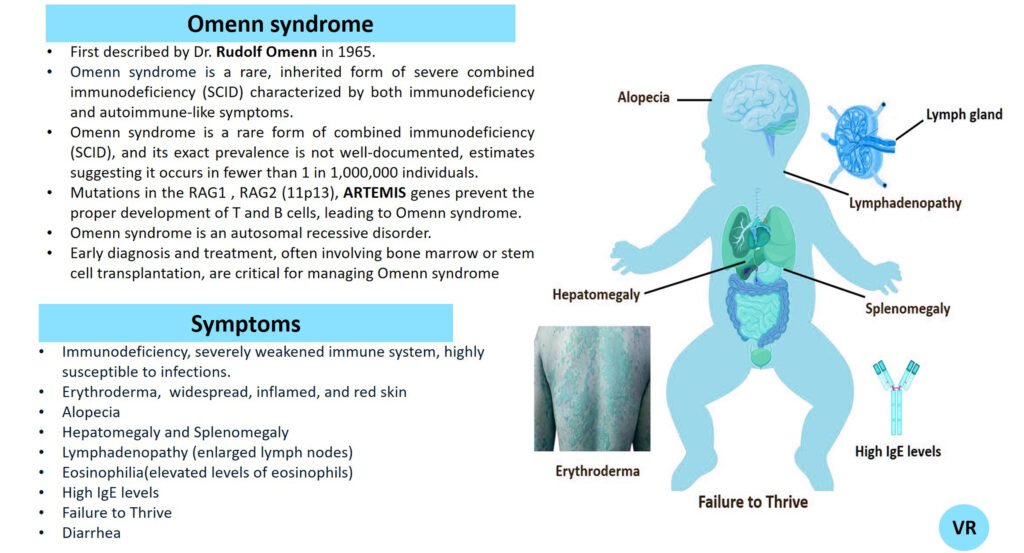
Severe Combined Immunodeficiency (SCID)
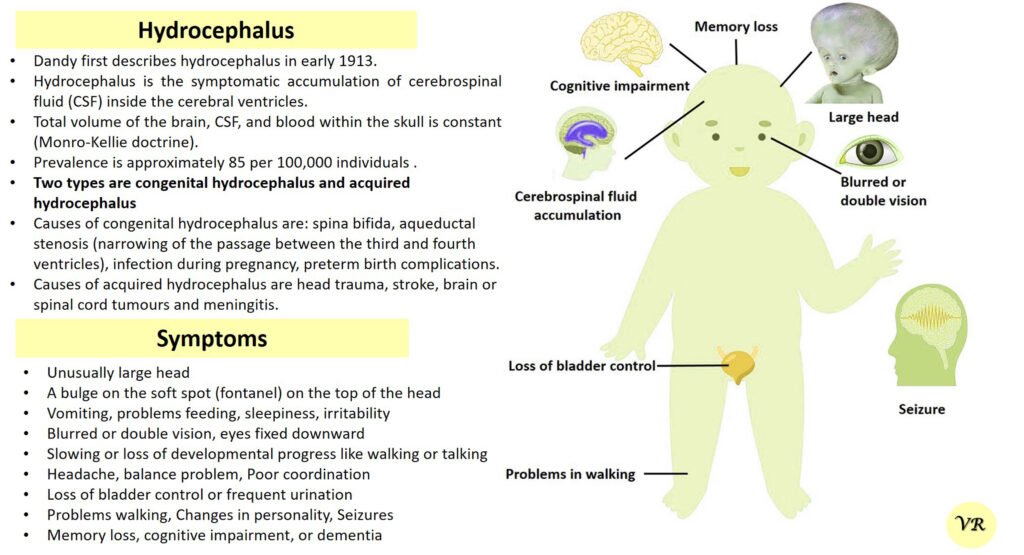
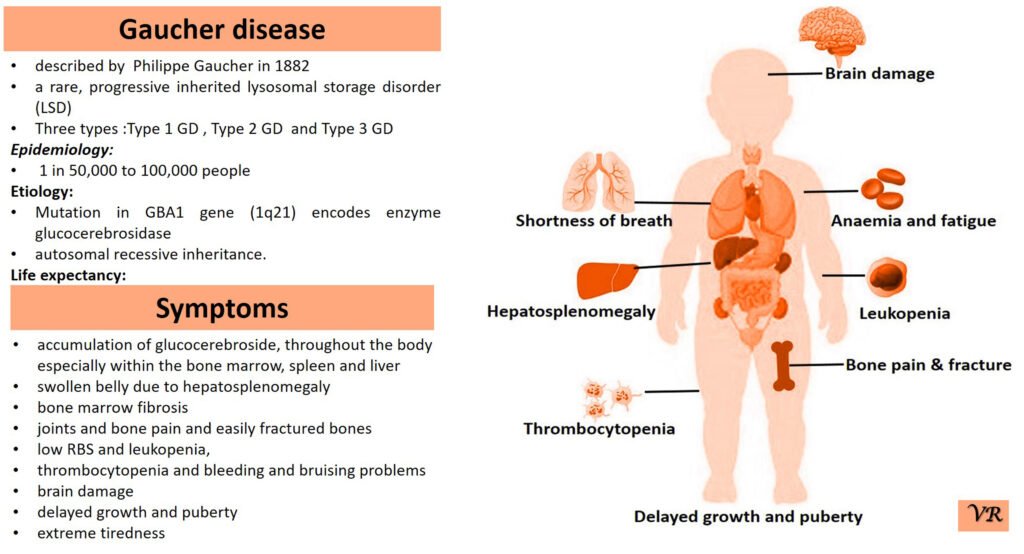
Severe Combined Immunodeficiency (SCID) is a group of rare, inherited disorders characterized by a severely compromised immune system. Individuals with SCID have little to no immune response due to profound defects in both T and B lymphocytes, the white blood cells essential for fighting infections. SCID is called “combined” because it affects both arms of the adaptive immune system: the cell-mediated response (T cells) and the humoral response (B cells). People with SCID have defects in both of these cell types, leading to a severely weakened immune response. SCID is estimated to occur in approximately 1 in 40,000 to 1 in 100,000 live births worldwide. X-linked SCID is the most common form of SCID and accounts for about 50-60% of all cases. X-linked SCID affects males primarily and occurs in about 1 in 50,000 to 1 in 100,000 male births. Symptoms of SCID include frequent severe infections (such as respiratory infections, ear infections, skin infections, fungal infections and digestive tract infections (e.g., diarrhea)), fail to grow or gain weight as expected, a condition called failure to thrive, lymphopenia, and delayed growth and development (infants may not meet developmental milestones). Severe Combined Immunodeficiency (SCID) can result from mutations in several different genes, all of which play crucial roles in the development and function of the immune system like- ADA (Adenosine Deaminase), RAG1 and RAG2 (Recombination Activating Genes 1 and 2), IL2RG (Interleukin-2 Receptor Gamma Chain), IL7R (Interleukin-7 Receptor Alpha), JAK3 (Janus Kinase 3), DCLRE1C (Artemis), and NHEJ1 (Non-Homologous End Joining 1, or Cernunnos) etc.
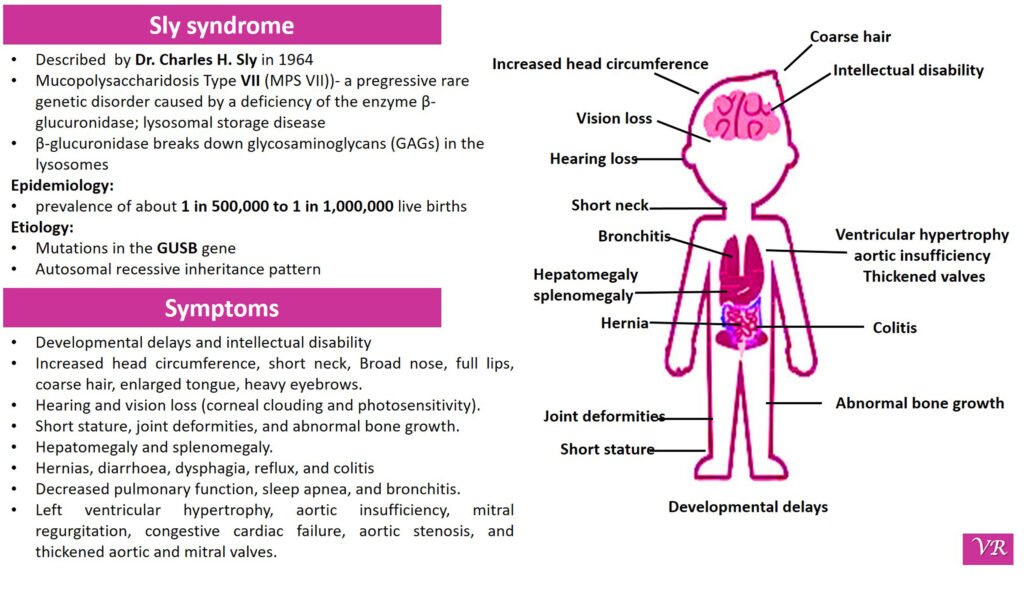
Sly syndrome
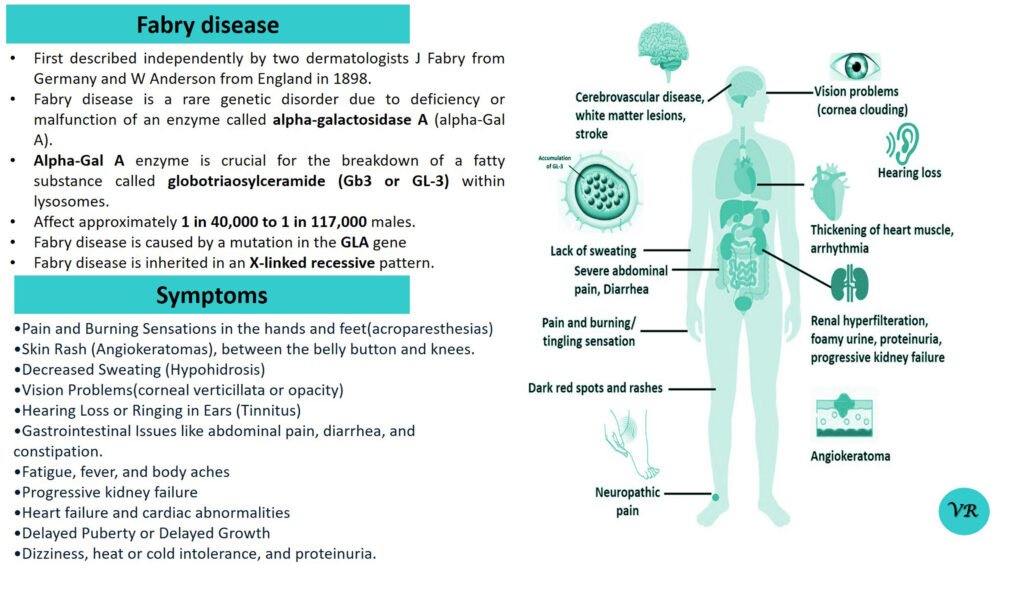
Sly syndrome (also known as β-glucuronidase deficiency or Mucopolysaccharidosis VII (MPS VII)) is a rare genetic disorder caused by a deficiency of the enzyme β-glucuronidase. This enzyme is crucial for breaking down certain complex carbohydrates called glycosaminoglycans (GAGs) in the lysosomes of cells. Without sufficient β-glucuronidase activity, these GAGs accumulate, leading to various clinical symptoms. The severity and timing of symptoms can vary depending on the individual, but common symptoms include developmental delays, broad nose, enlarged tongue, thick lips, prominent forehead, short stature, joint stiffness or contractures, abnormal bone growth, enlarged joints, hip dysplasia or dislocation , enlargement of the heart (cardiomegaly) , hearing loss (sensorineural hearing loss), corneal clouding (a clouding of the eye’s cornea), respiratory problems, hepatomegaly, splenomegaly, cognitive impairment and progressive neurological decline, thickening of the skin and connective tissues and recurrent infections. Sly syndrome is caused by mutations in the GUSB gene, which encodes the β-glucuronidase enzyme.
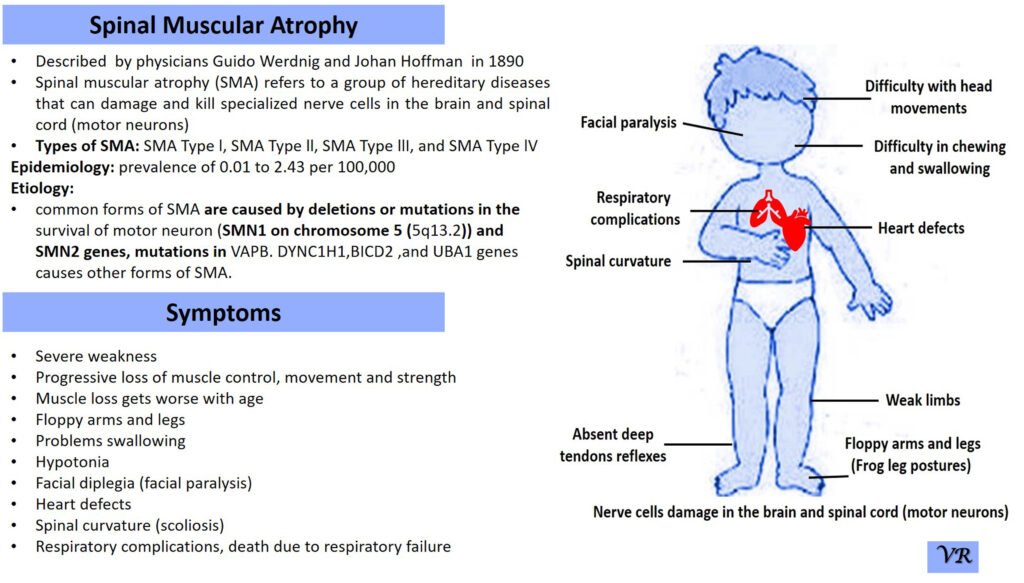
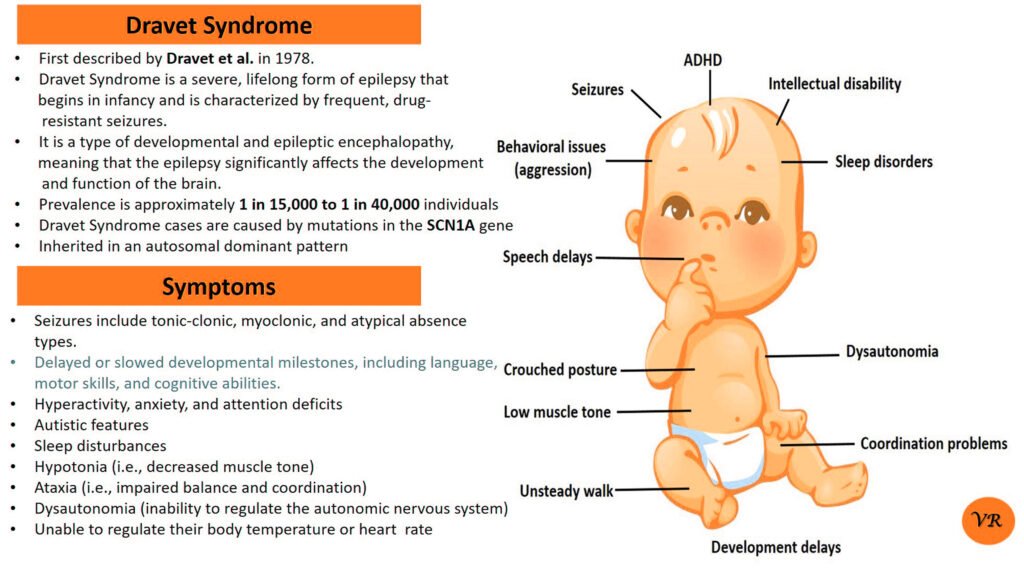
Spinal Muscular Atrophy (SMA)
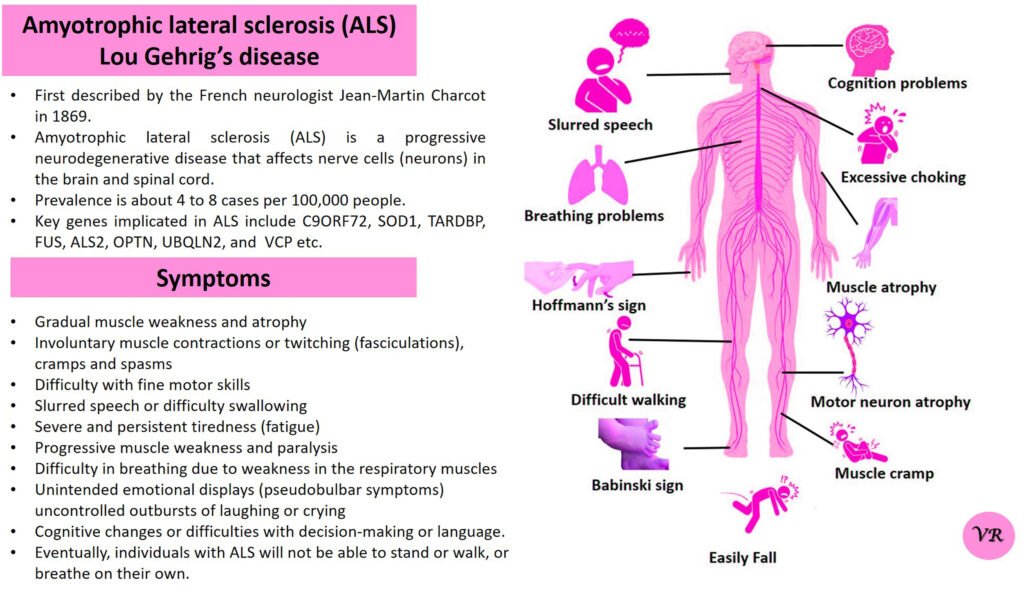
Spinal Muscular Atrophy (SMA) is a genetic disorder characterized by the progressive degeneration of motor neurons in the spinal cord and brainstem. These motor neurons are responsible for controlling voluntary muscle movements, such as crawling, walking, swallowing, and breathing. The loss of these neurons leads to muscle weakness and atrophy (wasting away) over time.
Different types of SMA have varying prevalences:
Type 1 (Werdnig-Hoffmann disease): This is the most common and severe form of SMA, accounting for about 50-60% of all cases.
Type 2: Represents approximately 20-30% of SMA cases.
Type 3 (Kugelberg-Welander disease): Accounts for about 10-20% of cases.
Type 4: This adult-onset form of SMA is rare and represents a small percentage of overall cases.
The severity and onset of symptoms can vary depending on the type of SMA, but common symptoms include:Muscle Weakness, Muscle Atrophy, Tremors,Hypotonia, Limited limited mobility, difficulty breathing, and swallowing.
Spinal Muscular Atrophy (SMA) is primarily caused by mutations in the Survival Motor Neuron 1 (SMN1) gene, but other genes can also be involved in the condition, contributing to different types and severities of SMA.
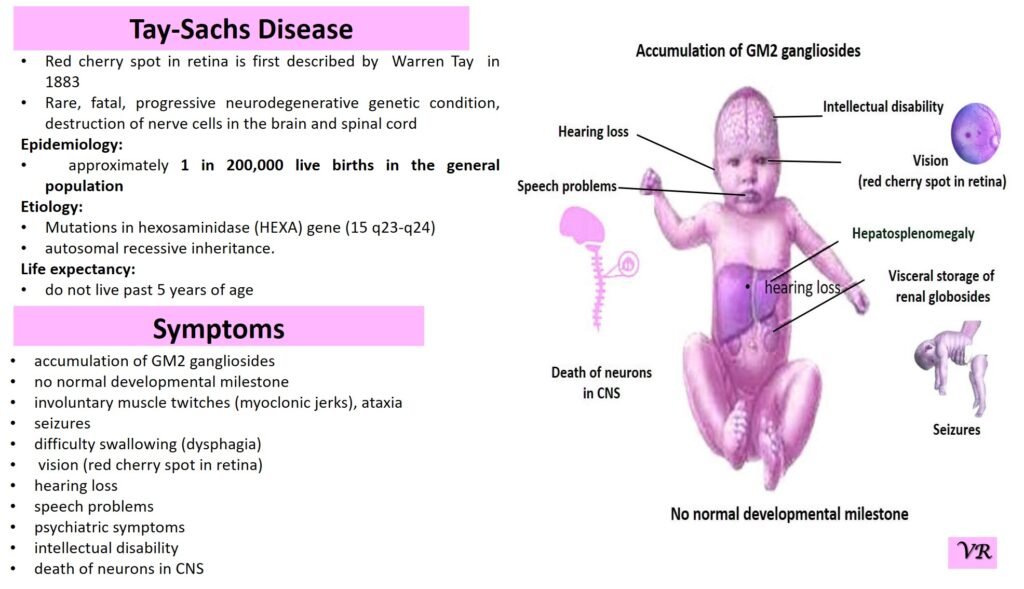
Tay-Sachs disease
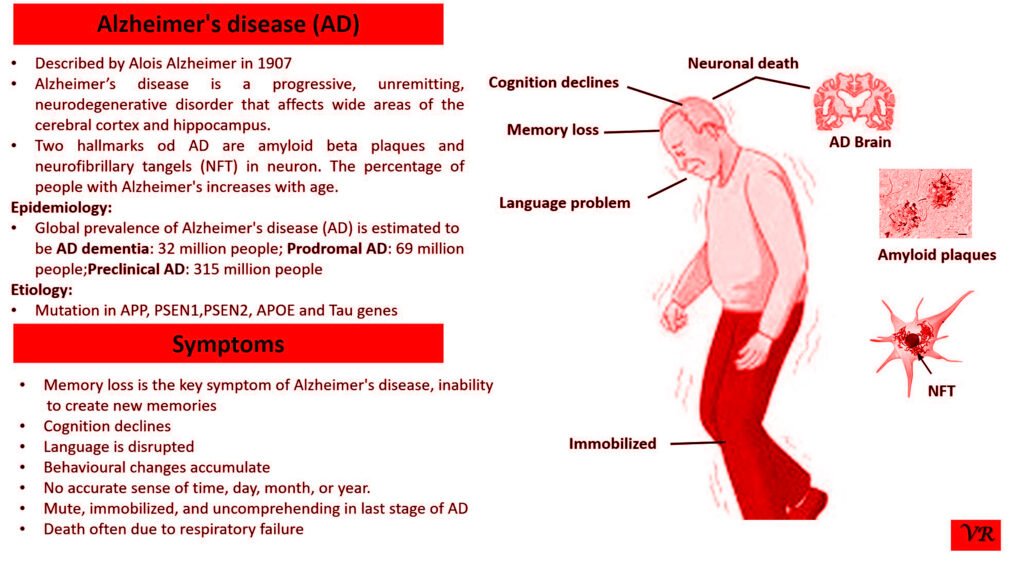
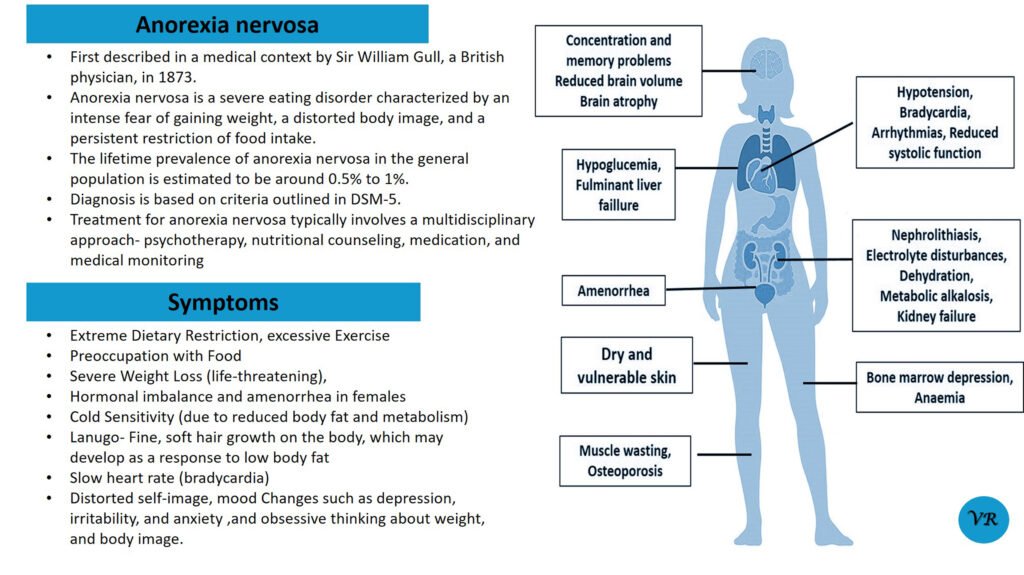
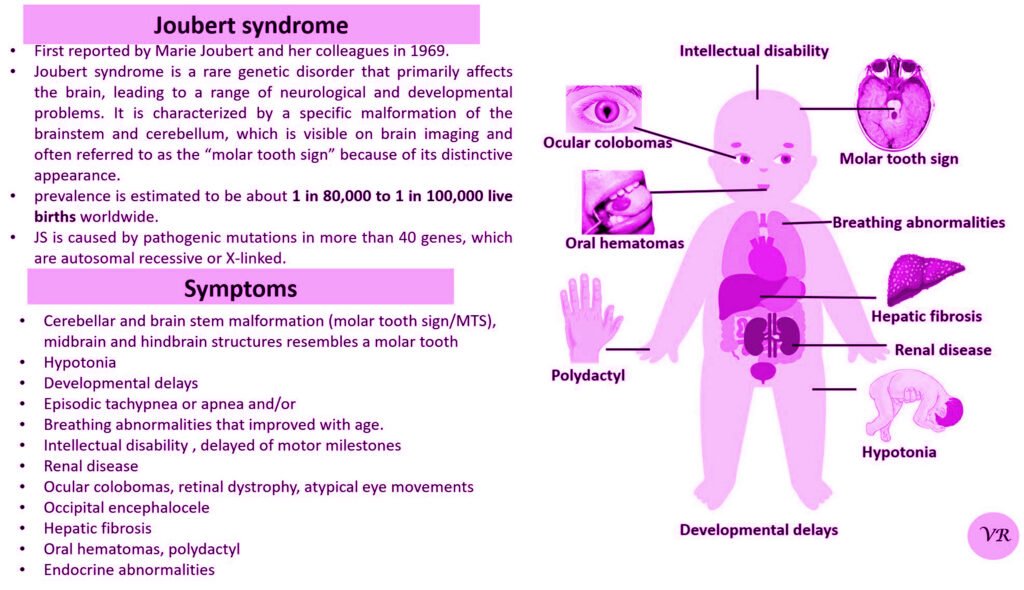
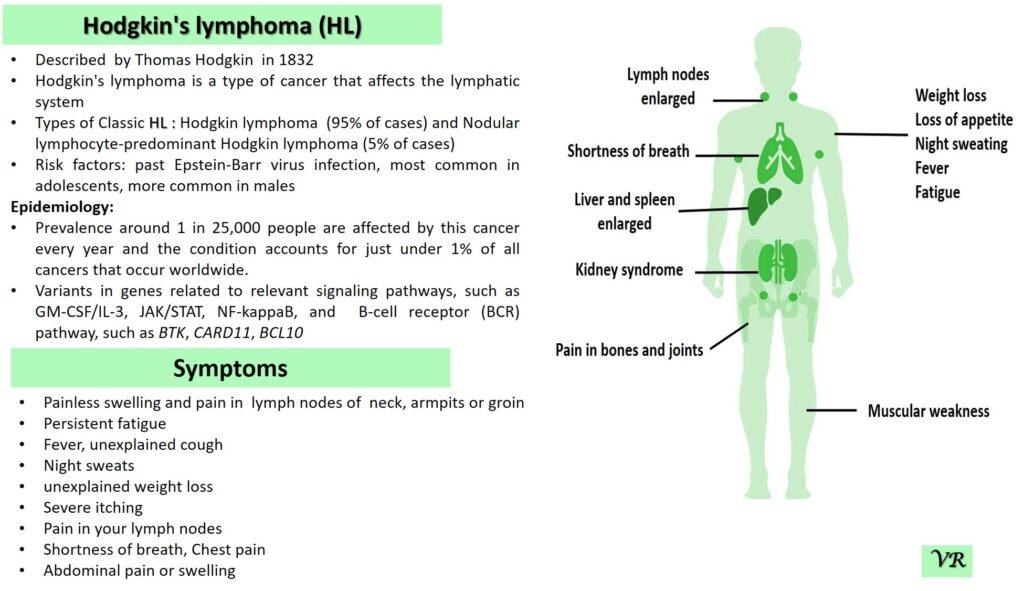
Tay-Sachs disease is a rare, inherited disorder that progressively destroys nerve cells (neurons) in the brain and spinal cord. It is a type of lysosomal storage disorder, meaning that it involves a defect in the body’s ability to break down specific lipids (fats) due to a deficiency of a particular enzyme. This disease is particularly known for its severe impact on infants and young children, leading to neurological decline and early death. Tay-Sachs disease affects approximately 1 in 320,000 to 1 in 400,000 live births worldwide. Tay-Sachs disease is categorized into several types based on the age of onset and severity of symptoms- Infantile Tay-Sachs Disease, Juvenile Tay-Sachs Disease, and Late-Onset Tay-Sachs Disease (LOTS). Symptoms of Tay-Sachs disease are motor and cognitive regression, severe developmental delays, vision and hearing loss, seizures, muscle weakness and paralysis, and psychiatric symptoms (like depression or personality changes). Cherry-red spot in the eye is a characteristic feature of Tay-Sachs disease. Tay-Sachs disease is caused by mutations in the hexosaminidase A (HEXA) gene, which is located on chromosome 15. The HEXA gene provides instructions for making a part of an enzyme called beta-hexosaminidase A.
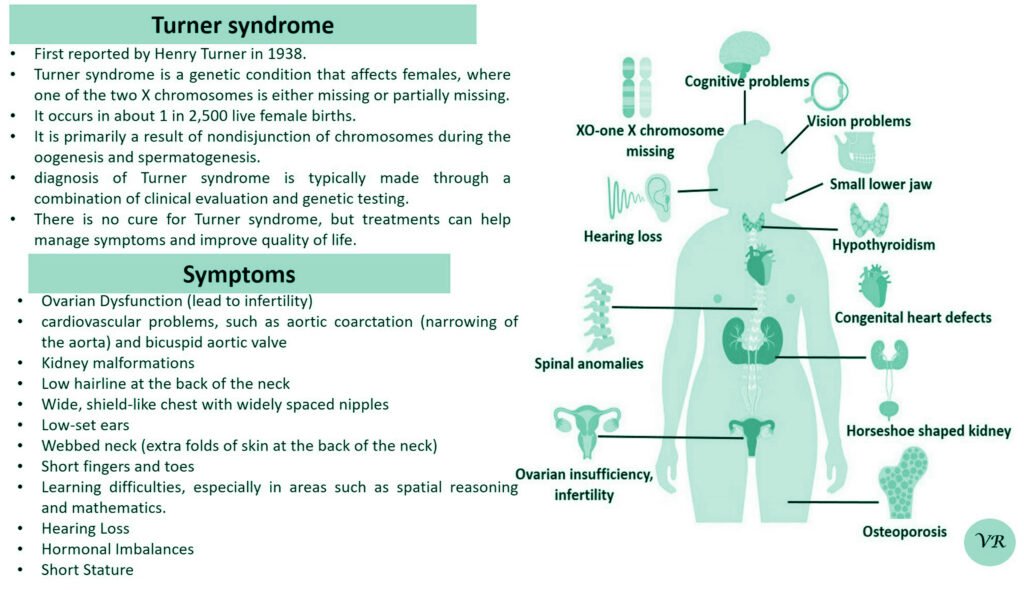
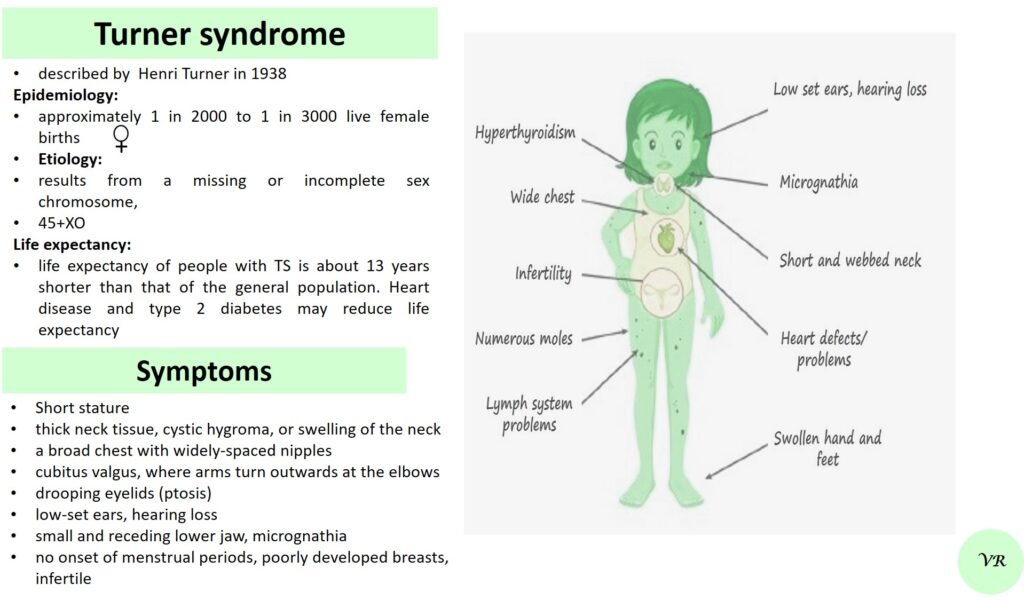
Turner syndrome
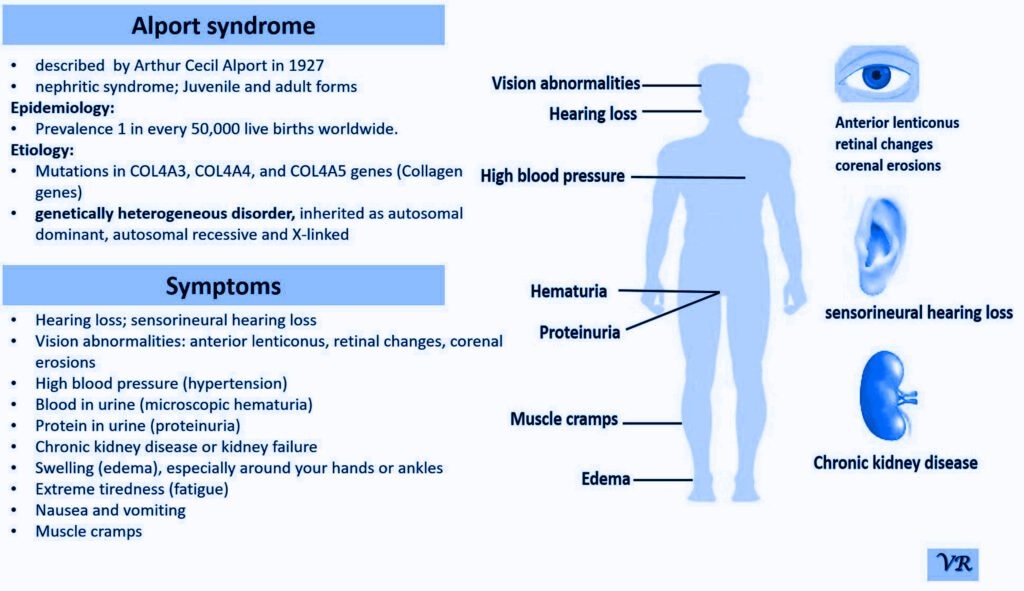
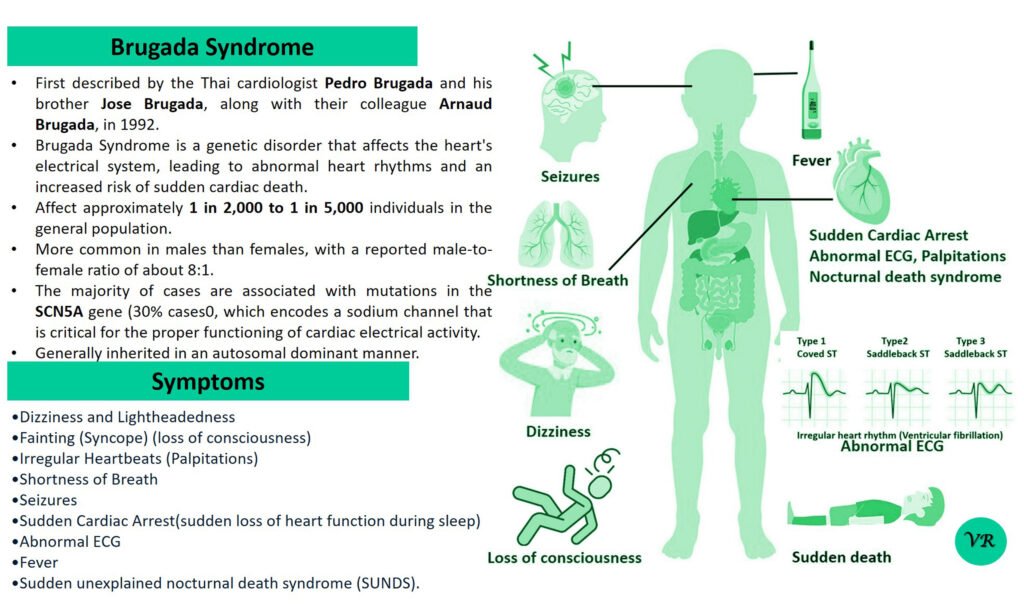
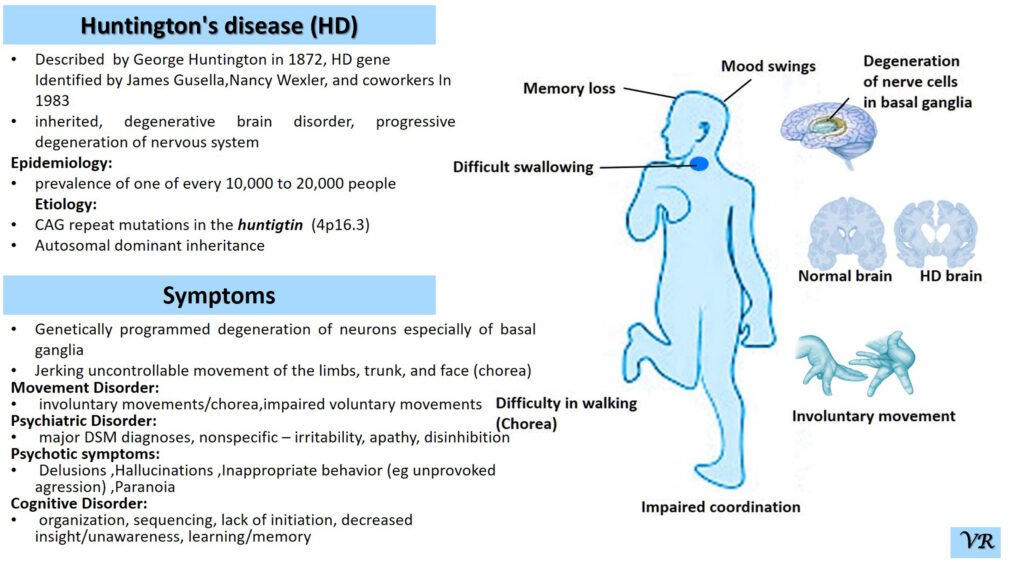
Turner syndrome is a genetic condition that affects females, where one of the two X chromosomes is either missing or partially missing. The prevalence of Turner syndrome is estimated to be about 1 in 2,000 – 3000 live female births and leads to a variety of physical, developmental, and medical issues. ymptoms of Turner syndrome are short stature, low hairline (at the back of the neck),wide chest with widely spaced nipples, low-set ears, webbed neck (extra folds of skin at the back of the neck), short fingers and toes , learning and developmental challenges, hearing loss, heart and kidney issues, ovarian dysfunction and hormonal imbalances (due to underdeveloped ovaries). The condition occurs when one of the two X chromosomes in females is missing or partially missing (22AA + XO), and it affects females of all racial and ethnic backgrounds. However, many cases are not immediately diagnosed, as some of the symptoms can be subtle or vary in severity, with some individuals being diagnosed later in life, often during adolescence or even adulthood.
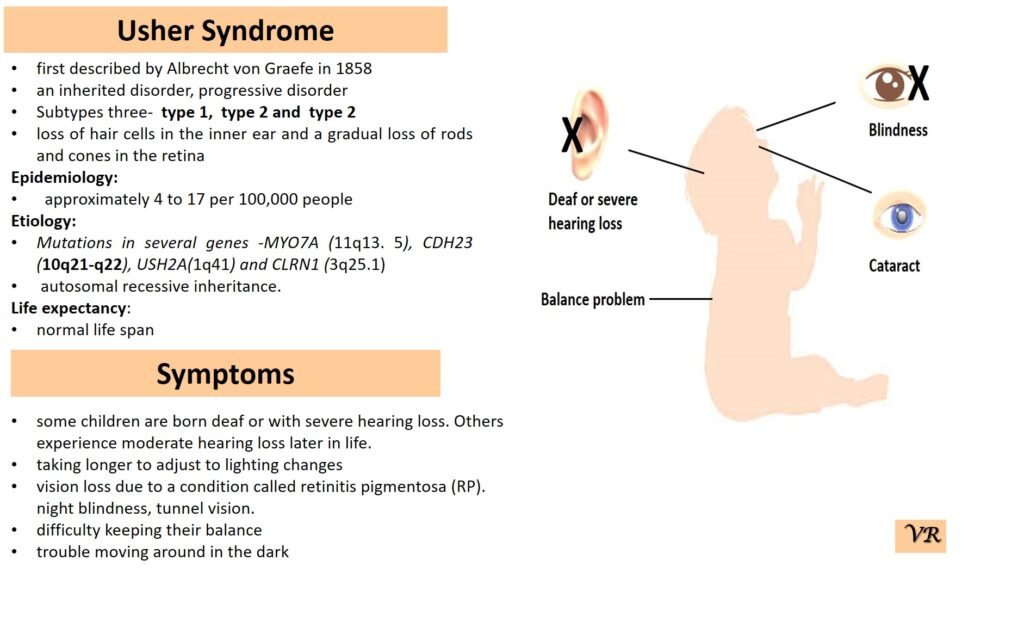
Usher syndrome
Usher syndrome is a genetic disorder characterized by a combination of hearing loss and vision impairment, and in some cases, balance problems. It is the most common condition that combines these two types of sensory deficits. Usher syndrome is classified into several types based on the severity and age of onset of symptoms. Symptoms of Usher Syndrome include hearing loss (congenital or early-onset hearing loss), vision loss (associated with retinitis pigmentosa and night blindness), and balance problems (due to vestibular dysfunction). Usher syndrome is inherited in an autosomal recessive manner. Different types of Usher syndrome are caused by mutations in different genes: Type 1 Usher syndrome ( MYO7A, USH2A, CDH23, and PCDH15), Type 2 Usher syndrome (USH2A gene), and Type 3 Usher syndrome (CLRN1, HARS, and USH3A). Myosin VIIA (Myo7A) gene is located at the long arm of chromosome 11 at position q13.4, consists of 49 exons. The size of MYO7A gene is 86.9 kb, encodes protein of 2215 amino acids.
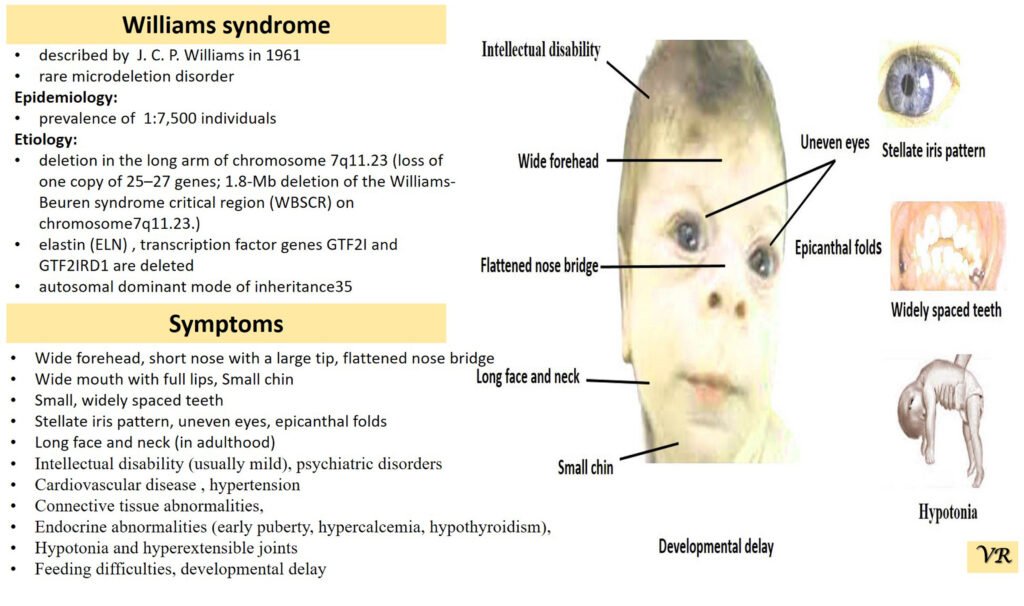
Williams syndrome
Williams syndrome is a rare genetic disorder characterized by a distinctive set of features, including cardiovascular problems, unique facial characteristics, developmental delays, and a specific personality profile. It results from a deletion of genetic material on chromosome 7, affecting several genes, including the ELN gene which encodes elastin. Symptoms of Williams syndrome are broad forehead, short nose with a broad tip, full cheeks, wide mouth, prominent chin, developmental delays (including motor and speech delays), cardiovascular issues (supravalvular aortic stenosis),mild to moderate intellectual disability,outgoing and friendly demeanor (charming and engaging personality),behavioral and psychological problems (anxiety, attention deficits, and hyperactivity), kidney problems, hearing loss, dental issues, and issues with fine motor skills. Williams syndrome is caused by a deletion of about 26-28 genes on chromosome 7, specifically in the region of 7q11.23. This deletion includes the ELN gene, which is crucial for the production of elastin, a protein that helps tissues return to their original shape after stretching.
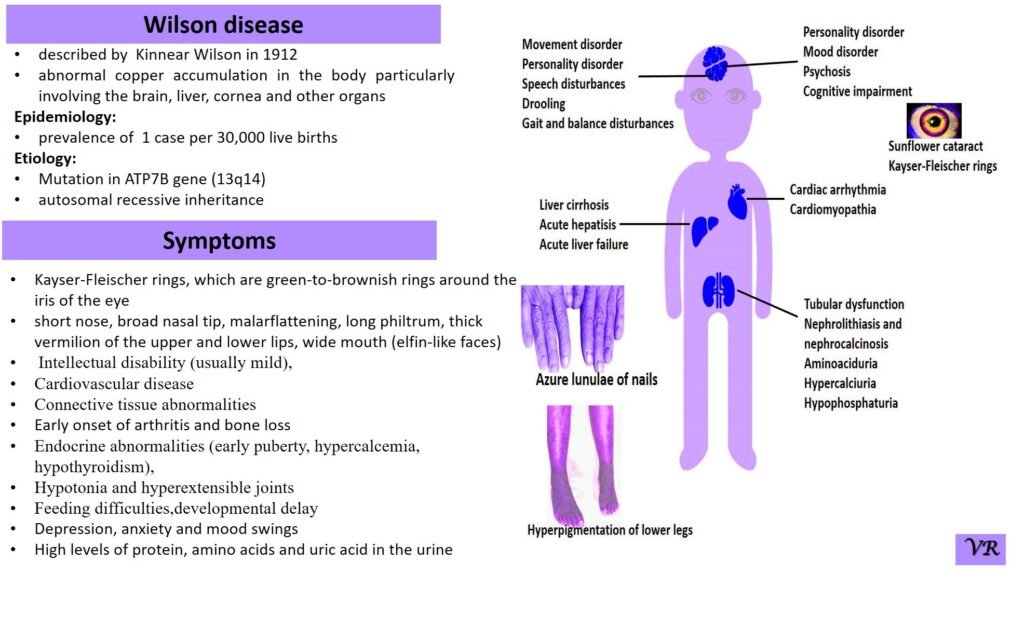
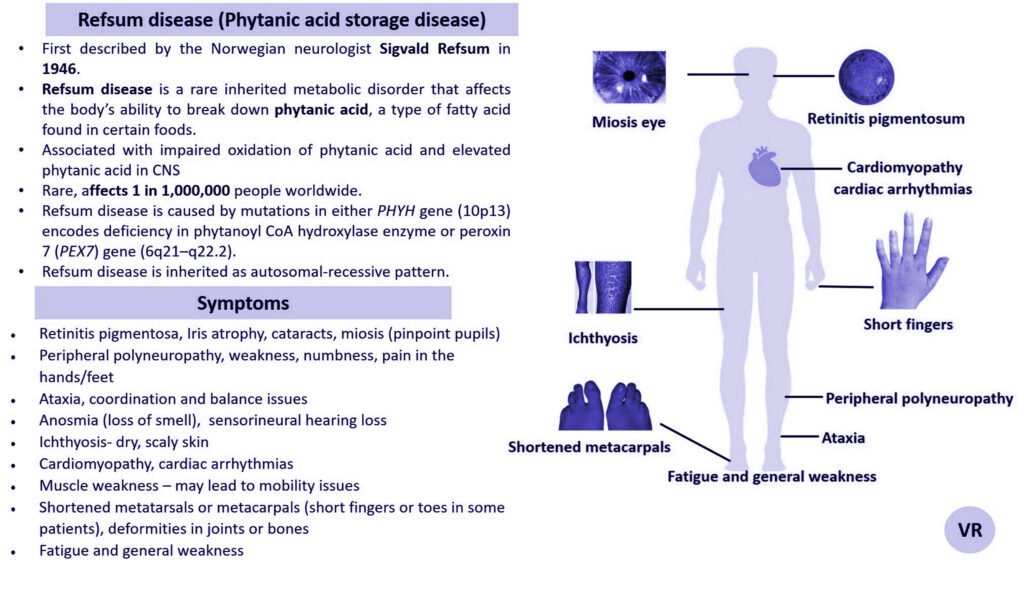
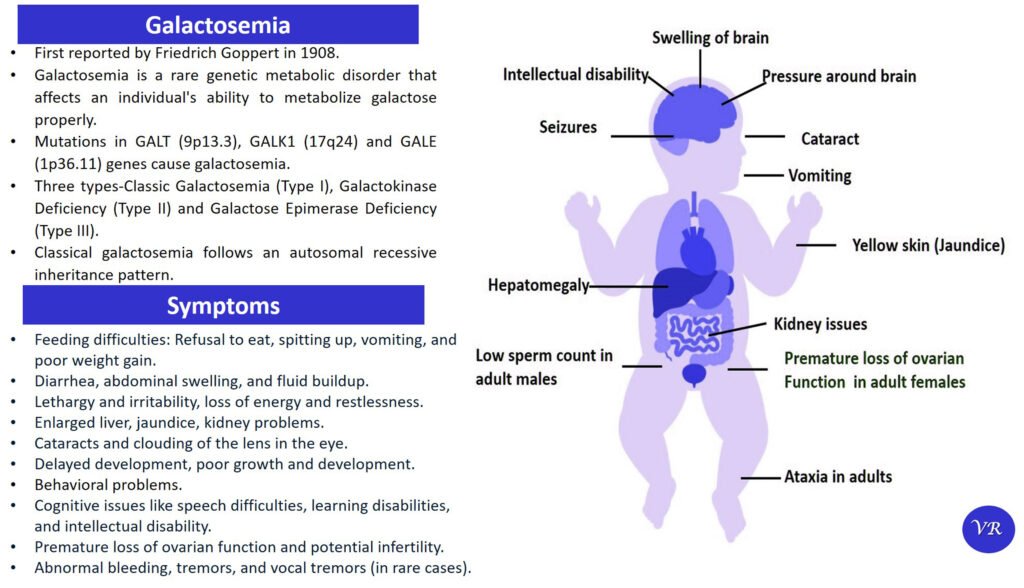
Wilson disease
Wilson disease is a rare genetic disorder characterized by the accumulation of copper in various tissues, particularly the liver and brain. This buildup of copper leads to oxidative damage and affects multiple organ systems, resulting in a range of symptoms. Wilson disease is estimated to occur in about 1 in 30,000 to 1 in 40,000 live births worldwide. Symptoms of Wilson disease are copper accumulation in the liver and brain, hepatomegaly, jaundice, abdominal pain, liver cirrhosis, tremors, dystonia (involuntary muscle contractions), dysarthria (speech difficulties), mood swings, and cognitive decline, golden-brown rings around the cornea of the eye (Kayser-Fleischer Rings), hematuria , proteinuria, and bone and joint problems. Wilson disease is caused by mutations in the ATP7B gene, and is inherited in an autosomal recessive manner.
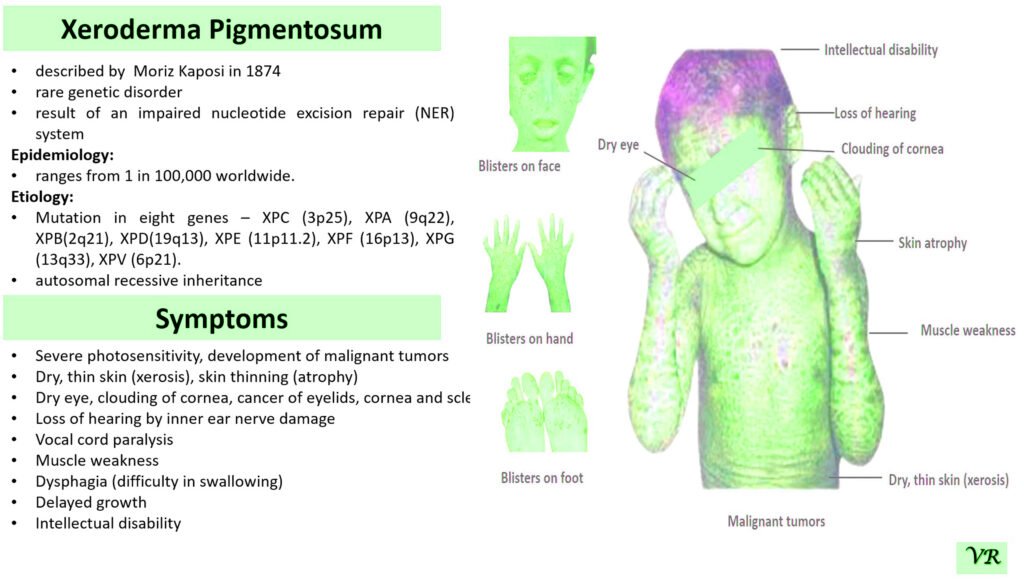
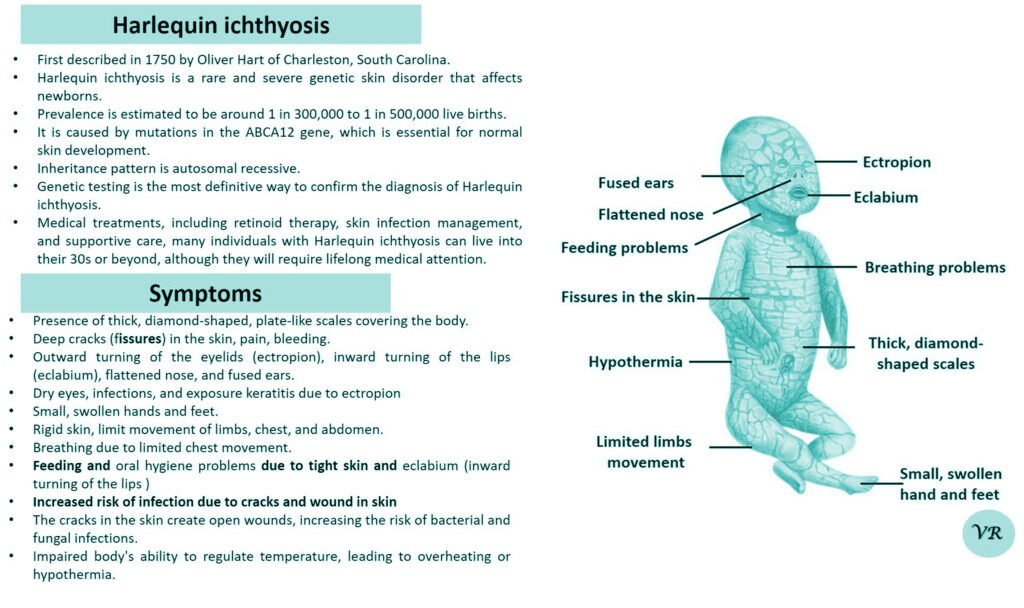
Xeroderma pigmentosum (XP)
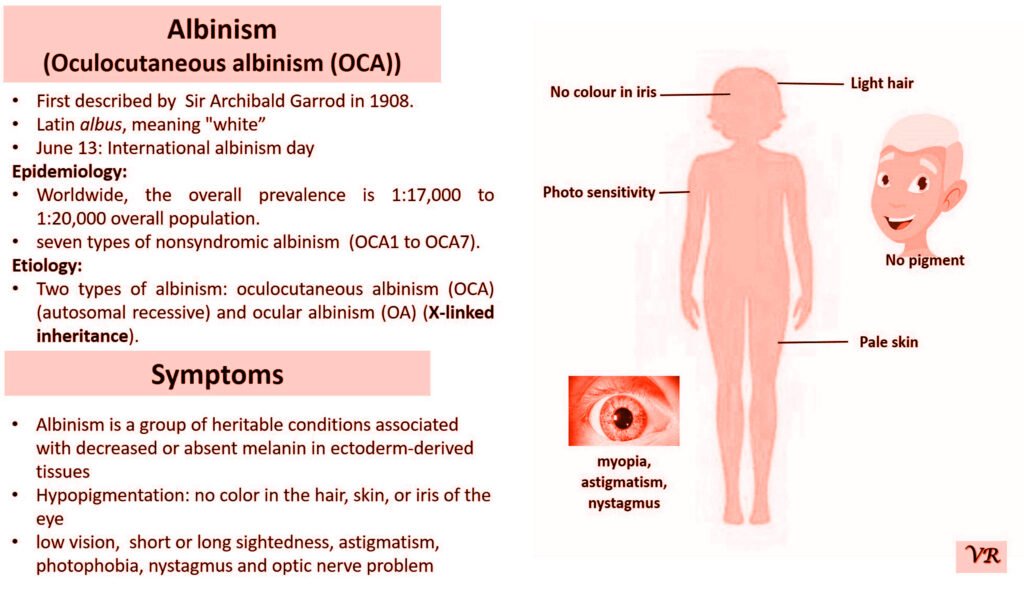
Xeroderma pigmentosum (XP) is a rare genetic disorder characterized by extreme sensitivity to ultraviolet (UV) light, leading to severe skin damage and a high risk of skin cancers. The condition results from defects in the DNA repair mechanisms that normally correct UV-induced damage to the skin’s DNA. XP is caused by mutations in genes responsible for nucleotide excision repair (NER), a critical process for repairing UV-induced DNA damage. The most common genes associated with XP include: XRCC1,XPA, XPC, XPE, XPF, and XPG, etc. Each gene defect leads to a specific XP subtype, with varying degrees of severity and associated symptoms. XP is inherited in an autosomal recessive manner. XRCC1 gene is located on the long arm of chromosome 19 at position 13.2 and consists of 17 exons. Size of gene is 31.9 kb and encodes a protein of 633 amino acids.